Glycogen storage disease type I
Glycogen storage disease type I (GSD I) is an inherited disease that prevents the liver from properly breaking down stored glycogen, which is necessary in maintain adequate blood sugar levels. GSD I is divided into two main types, GSD Ia and GSD Ib, which differ in cause, presentation, and treatment. There are also possibly rarer subtypes, the translocases for inorganic phosphate (GSD Ic) or glucose (GSD Id); however, a recent study suggests that the biochemical assays used to differentiate GSD Ic and GSD Id from GSD Ib are not reliable, and are therefore GSD Ib.[1]
| GSD Type I | |
|---|---|
| Other names | von Gierke disease |
| Symbol for Glycogen Storage Disease Type I | |
| Pronunciation |
|
| Specialty | Endocrinology, genetics, hematology, immunology |
| Complications | Lactic acidosis, hyperlipidemia, non-alcoholic fatty liver disease, hepatocellular adenoma, inflammatory bowel disease |
| Duration | Lifetime |
| Types | Type Ia, type Ib |
| Causes | Autosomal recessive inheritance |
| Diagnostic method | Genetic testing, hypoglycemia, hepatomegaly Type Ib: neutropenia |
| Treatment | Cornstarch, diet |
| Medication | Filgrastim |
| Frequency | 1 in 100,000 live births |
GSD Ia is caused by a deficiency in the enzyme glucose-6-phosphatase; GSD Ib, a deficiency in the transport protein glucose-6-phosphate translocase. Because glycogenolysis is the principal metabolic mechanism by which the liver supplies glucose to the body during fasting, both deficiencies cause severe hypoglycemia and, over time, excess glycogen storage in the liver and (in some cases) in the kidneys.
Because of the glycogen buildup, GSD I patients typically present with enlarged livers from non-alcoholic fatty liver disease.[2] Other functions of the liver and kidneys are initially intact in GSD I, but are susceptible to other problems. Without proper treatment, GSD I causes chronic low blood sugar, which can lead to excessive lactic acid, and abnormally high lipids in the blood, and other problems. Frequent feedings of cornstarch or other carbohydrates are the principal treatment for all forms of GSD I.
GSD Ib also features chronic neutropenia due to a dysfunction in the production of neutrophils in the bone marrow. This immunodeficiency, if untreated, makes GSD Ib patients susceptible to infection.[3] The principal treatment for this feature of GSD Ib is filgrastim; however, patients often still require treatment for frequent infections, and a chronically enlarged spleen is a common side effect.[4] GSD Ib patients often present with inflammatory bowel disease.[5]
It is the most common of the glycogen storage diseases. GSD I has an incidence of approximately 1 in 100,000 births in the American population, and approximately 1 in 20,000 births among Ashkenazi Jews.[6] The disease was named after German doctor Edgar von Gierke, who first described it in 1929.[7][8]
Signs and symptoms
Early research into GSD I identified numerous clinical manifestations falsely thought to be primary features of the genetic disorder. However, continuing research has revealed that these clinical features are the consequences of only one (in GSD Ia) or two (in GSD Ib) fundamental abnormalities:
- impairment in the liver's ability to convert stored glycogen into glucose through glycogenolysis[9]
- in GSD Ib, impairment of the neutrophil's ability to take up glucose, resulting in neutrophil dysfunction and neutropenia[10]
These fundamental abnormalities give rise to a small number of primary clinical manifestations, which are the features considered in diagnosis of GSD I:
- Low blood sugar (hypoglycemia), due to impairment of glycogen breakdown (glycogenolysis) causing insufficient fasting blood glucose[11]
- hepatomegaly of non-alcoholic fatty liver disease, due to impairment of glycogenolysis causing glycogen accumulation in the liver[2]
- in GSD Ib, increased infection risk, due to neutropenia and neutrophil dysfunction[3]
Affected people commonly present with secondary clinical manifestations, linked to one or more of the primary clinical manifestations:
- High levels of uric acid in the blood and attendant risk of gout or kidney damage, caused by low serum insulin levels in prolonged hypoglycemia
- High levels of lactic acid in the blood, in extreme cases leading to lactic acidosis, caused by prolonged hypoglycemia[11]
- hepatic adenomas developing in adulthood[12] and attendant risk of anemia,[13] suspected to be caused by blood glucose dysregulation in the presence of non-alcoholic fatty liver disease
- in GSD Ib, inflammatory bowel disease and attendant risk of anemia,[13] caused by neutrophil dysfunction and exacerbated by the increased carbohydrate intake required to prevent hypoglycemia[3]
In addition, there are several clinical manifestations that often result from the treatment of the primary clinical manifestations:
- pancreatic hypertrophy, due to increased carbohydrate intake causing frequent engagement of the insulin response[14]
- in GSD Ib, splenomegaly, due to the long-term use of filgrastim to treat neutropenia causing sequestration of blood factors in the spleen[4]
- in GSD Ib, an abnormally low number of platelets in the blood may occur, due to long-term use of filgrastim causing sequestration of platelets in the spleen[15]
- in GSD Ib, anemia, due to long-term use of filgrastim causing sequestration of hemoglobin in the spleen, potentially exacerbated by uncontrolled inflammatory bowel disease[16]
Hypoglycemia
Low blood sugar (hypoglycemia) is the primary clinical symptom common to both GSD Ia and GSD Ib and most often prompts initial diagnosis of the disease. During fetal development in utero, maternal glucose transferred across the placenta prevents hypoglycemia. However, after birth, the inability to maintain blood glucose from stored glycogen in the liver causes measurable hypoglycemia in no more than 1–2 hours after feedings. Without proper dietary treatment after birth, prolonged hypoglycemia often leads to sudden lactic acidosis that can induce primary respiratory distress in the newborn period, as well as ketoacidosis.
Neurological manifestations of hypoglycemia are less severe in GSD I than in other instances. Rather than acute hypoglycemia, GSD I patients experience persistent mild hypoglycemia. The diminished likelihood of neurological manifestations is due to the habituation of the brain to mild hypoglycemia. Given the reduced blood glucose level, the brain adapts to using alternative fuels like lactate. These gradual metabolic adaptations during infancy make severe symptoms like unconsciousness or seizure uncommon before diagnosis.
In the early weeks of life, undiagnosed infants with GSD I tolerate persistent hypoglycemia and compensated lactic acidosis between feedings without symptoms. Without consistent carbohydrate feeding, infant blood glucose levels typically measure between 25 and 50 mg/dL (1.4 to 2.8 mmol/L). After weeks to months without treatment with consistent oral carbohydrates, infants will progress to show clear symptoms of hypoglycemia and lactic acidosis. Infants may present with paleness, clamminess, irritability, respiratory distress, and an inability to sleep through the night even in the second year of life. Developmental delay is not an intrinsic effect of GSD I, but is common if the diagnosis is not made in early infancy.
Genetics

GSD I is inherited in an autosomal recessive manner. People with one copy of the faulty gene are carriers of the disease and have no symptoms. As with other autosomal recessive diseases, each child born to two carriers of the disease has a 25% chance of inheriting both copies of the faulty gene and manifesting the disease. Unaffected parents of a child with GSD I can be assumed to be carriers. Prenatal diagnosis has been made by fetal liver biopsy at 18–22 weeks of gestation, but no fetal treatment has been proposed. Prenatal diagnosis is possible with fetal DNA obtained by chorionic villus sampling when a fetus is known to be at risk.
The most common forms of GSD I are designated GSD Ia and GSD Ib, the former accounting for over 80% of diagnosed cases and the latter for less than 20%. A few rarer forms have been described.
- GSD Ia results from mutations of G6PC, the gene for glucose-6-phosphatase,[17] located on chromosome 17q21.[18]
- GSD Ib results from mutations of the gene for SLC37A4 or "G6PT1", the glucose-6-phosphate transporter.[18][19]
- GSD Ic results from mutations of SLC17A3 or SLC37A4.[20]
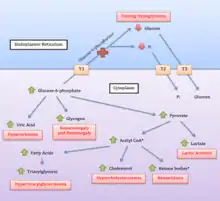
Glucose-6-phosphatase is an enzyme located on the inner membrane of the endoplasmic reticulum. The catalytic unit is associated with a calcium binding protein, and three transport proteins (T1, T2, T3) that facilitate movement of glucose-6-phosphate (G6P), phosphate, and glucose (respectively) into and out of the enzyme.
Pathophysiology
Normal carbohydrate balance and maintenance of blood glucose levels
Glycogen in liver and (to a lesser degree) kidneys serves as a form of stored, rapidly accessible glucose, so that the blood glucose level can be maintained between meals. For about 3 hours after a carbohydrate-containing meal, high insulin levels direct liver cells to take glucose from the blood, to convert it to glucose-6-phosphate (G6P) with the enzyme glucokinase, and to add the G6P molecules to the ends of chains of glycogen (glycogen synthesis). Excess G6P is also shunted into production of triglycerides and exported for storage in adipose tissue as fat.
When digestion of a meal is complete, insulin levels fall, and enzyme systems in the liver cells begin to remove glucose molecules from strands of glycogen in the form of G6P. This process is termed glycogenolysis. The G6P remains within the liver cell unless the phosphate is cleaved by glucose-6-phosphatase. This dephosphorylation reaction produces free glucose and free PO
4 anions. The free glucose molecules can be transported out of the liver cells into the blood to maintain an adequate supply of glucose to the brain and other organs of the body. Glycogenolysis can supply the glucose needs of an adult body for 12–18 hours.
When fasting continues for more than a few hours, falling insulin levels permit catabolism of muscle protein and triglycerides from adipose tissue. The products of these processes are amino acids (mainly alanine), free fatty acids, and lactic acid. Free fatty acids from triglycerides are converted to ketones, and to acetyl-CoA. Amino acids and lactic acid are used to synthesize new G6P in liver cells by the process of gluconeogenesis. The last step of normal gluconeogenesis, like the last step of glycogenolysis, is the dephosphorylation of G6P by glucose-6-phosphatase to free glucose and PO
4.
Thus glucose-6-phosphatase mediates the final, key, step in both of the two main processes of glucose production during fasting. The effect is amplified because the resulting high levels of glucose-6-phosphate inhibit earlier key steps in both glycogenolysis and gluconeogenesis.
Pathophysiology
The principal metabolic effects of deficiency of glucose-6-phosphatase are hypoglycemia, lactic acidosis, hypertriglyceridemia, and hyperuricemia.
The hypoglycemia of GSD I is termed "fasting", or "post-absorptive", usually about 4 hours after the complete digestion of a meal. This inability to maintain adequate blood glucose levels during fasting results from the combined impairment of both glycogenolysis and gluconeogenesis. Fasting hypoglycemia is often the most significant problem in GSD I, and typically the problem that leads to the diagnosis. Chronic hypoglycemia produces secondary metabolic adaptations, including chronically low insulin levels and high levels of glucagon and cortisol.
Lactic acidosis arises from impairment of gluconeogenesis. Lactic acid is generated both in the liver and muscle and is oxidized by NAD+ to pyruvic acid and then converted via the gluconeogenic pathway to G6P. Accumulation of G6P inhibits conversion of lactate to pyruvate. The lactic acid level rises during fasting as glucose falls. In people with GSD I, it may not fall entirely to normal even when normal glucose levels are restored.
Hypertriglyceridemia resulting from amplified triglyceride production is another indirect effect of impaired gluconeogenesis, amplified by chronically low insulin levels. During fasting, the normal conversion of triglycerides to free fatty acids, ketones, and ultimately acetyl-CoA is impaired. Triglyceride levels in GSD I can reach several times normal and serve as a clinical index of "metabolic control".
Hyperuricemia results from a combination of increased generation and decreased excretion of uric acid, which is generated when increased amounts of G6P are metabolized via the pentose phosphate pathway. It is also a byproduct of purine degradation. Uric acid competes with lactic acid and other organic acids for renal excretion in the urine. In GSD I increased availability of G6P for the pentose phosphate pathway, increased rates of catabolism, and diminished urinary excretion due to high levels of lactic acid all combine to produce uric acid levels several times normal. Although hyperuricemia is asymptomatic for years, kidney and joint damage gradually accrue.
Elevated lactate and lactic acidosis
High levels of lactic acid in the blood are observed in all people with GSD I, due to impaired gluconeogenesis. Baseline elevations generally range from 4 to 10 mol/mL, which will not cause any clinical impact. However, during and after an episode of low blood sugar, lactate levels will abruptly rise to exceed 15 mol/mL, the threshold for lactic acidosis. Symptoms of lactic acidosis include vomiting and hyperpnea, both of which can exacerbate hypoglycemia in the setting of GSD I. In cases of acute lactic acidosis, patients need emergency care to stabilize blood oxygen, and restore blood glucose. Proper identification of lactic acidosis in undiagnosed children presents a challenge, since the first symptoms are typically vomiting and dehydration, both of which mimic childhood infections like gastroenteritis or pneumonia. Moreover, both of these common infections can precipitate more severe hypoglycemia in undiagnosed children, making diagnosis of the underlying cause difficult.
As elevated lactate persists, uric acid, ketoacids, and free fatty acids further increase the anion gap. In adults and children, the high concentrations of lactate cause significant discomfort in the muscles. This discomfort is an amplified form of the burning sensation a runner may feel in the quadriceps after sprinting, which is caused by a brief buildup of lactic acid. Proper control of hypoglycemia in GSD I eliminates the possibility for lactic acidosis.
Elevated urate and complications
High levels of uric acid often present as a consequence of elevated lactic acid in GSD I patients. When lactate levels are elevated, blood-borne lactic acid competes for the same kidney tubular transport mechanism as urate, limiting the rate that urate can be cleared by the kidneys into the urine. If present, increased purine catabolism is an additional contributing factor. Uric acid levels of 6 to 12 mg/dl (530 to 1060 umol/L) are common among GSD I patients, if the disease is not properly treated. In some affected people, the use of the medication allopurinol is necessary to lower blood urate levels. Consequences of hyperuricemia among GSD I patients include the development of kidney stones and the accumulation of uric acid crystals in joints, leading to kidney disease and gout, respectively.
Hyperlipidemia and plasma effects
Elevated triglycerides in GSD I result from low serum insulin in patients with frequent prolonged hypoglycemia. It may also be caused by intracellular accumulation of glucose-6-phosphate with secondary shunting to pyruvate, which is converted into Acetyl-CoA, which is transported to the cytosol where the synthesis of fatty acids and cholesterol occurs. Triglycerides above the 3.4 mmol/L (300 mg/dL) range may produce visible lipemia, and even a mild pseudohyponatremia due to a reduced aqueous fraction of the blood plasma. In GSD I, cholesterol is typically only mildly elevated compared to other lipids.
Hepatomegaly
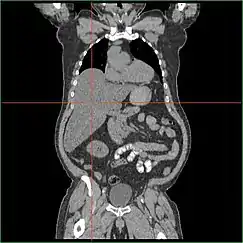
Impairment in the liver's ability to perform gluconeogenesis leads to clinically apparent hepatomegaly. Without this process, the body is unable to liberate glycogen from the liver and convert it into blood glucose, leading to an accumulation of stored glycogen in the liver. Hepatomegaly from the accumulation of stored glycogen in the liver is considered a form of non-alcoholic fatty liver disease. GSD I patients present with a degree of hepatomegaly throughout life, but severity often relates to the consumption of excess dietary carbohydrate. Reductions in the mass of the liver are possible, since most patients retain residual hepatic function that allows for the liberation of stored glycogen at a limited rate.
GSD I patients often present with hepatomegaly from the time of birth. In fetal development, maternal glucose transferred to the fetus prevents hypoglycemia, but the storage of glucose as glycogen in the liver leads to hepatomegaly. There is no evidence that this hepatomegaly presents any risk to proper fetal development.
Hepatomegaly in GSD type I generally occurs without sympathetic enlargement of the spleen. GSD Ib patients may present with splenomegaly, but this is connected to the use of filgrastim to treat neutropenia in this subtype, not comorbid hepatomegaly. Hepatomegaly will persist to some degree throughout life, often causing the abdomen to protrude, and in severe cases may be palpable at or below the navel. In GSD-related non-alcoholic fatty liver disease, hepatic function is usually spared, with liver enzymes and bilirubin remaining within the normal range. However, liver function may be affected by other hepatic complications in adulthood, including the development of hepatic adenomas.
Hepatic adenomas
The specific etiology of hepatic adenomas in GSD I remains unknown, despite ongoing research. The typical GSD I patient presenting with at least one adenoma is an adult, though lesions have been observed in patients as young as fourteen. Adenomas, composed of heterogeneous neoplasms, may occur individually or in multiples. Estimates on the rate of conversion of a hepatocellular adenoma into hepatocellular carcinoma in GSD I range from 0% to 11%, with the latter figure representing more recent research. One reason for the increasing estimate is the growing population of GSD I patients surviving into adulthood, when most adenomas develop.
Treatment standards dictate regular observation of the liver by MRI or CT scan to monitor for structural abnormalities. Hepatic adenomas may be misidentified as focal nodular hyperplasia in diagnostic imaging, though this condition is rare. However, hepatic adenomas in GSD I uniquely involve diffuse Mallory hyaline deposition, which is otherwise commonly observed in focal nodular hyperplasia. Unlike common hepatic adenomas related to oral contraception, hemorrhaging in GSD I patients is rare.
While the reason for the high prevalence of adenomas in GSD I is unclear, research since the 1970s has implicated serum glucagon as a potential driver. In studies, patients that have been put on a dietary regimen to keep blood sugar in a normal range spanning 72 to 108 mg/dL (4.0 to 6.0 mmol/L) have shown a decreased likelihood of developing adenomas. Moreover, patients with well controlled blood glucose have consistently seen a reduction in the size and number of hepatic adenomas, suggesting that adenomas may be caused by imbalances of hepatotropic agents like serum insulin and especially serum glucagon in the liver.[21]
Osteopenia
Patients with GSD I will often develop osteopenia. The specific etiology of low bone mineral density in GSD is not known, though it is strongly associated with poor metabolic control. Osteopenia may be directly caused by hypoglycemia, or the resulting endocrine and metabolic sequelae. Improvements in metabolic control have consistently been shown to prevent or reverse clinically relevant osteopenia in GSD I patients.[22] In cases where osteopenia progresses with age, bone mineral density in the ribs is typically more severe than in the vertebrae.[23] In some cases bone mineral density T-score will drop below -2.5, indicating osteoporosis. There is some evidence that osteopenia may be connected with associated kidney abnormalities in GSD I, particularly glomular hyperfiltration.[24] The condition also seems responsive to calcium supplementation. In many cases bone mineral density can increase and return to the normal range given proper metabolic control and calcium supplementation alone, reversing osteopenia.
Kidney effects
The kidneys are usually 10 to 20% enlarged with stored glycogen. In adults with GSD I, chronic glomerular damage similar to diabetic nephropathy may lead to kidney failure. GSD I may present with various kidney complications. Renal tubular abnormalities related to hyperlactatemia are seen early in life, likely because prolonged lactic acidosis is more likely to occur in childhood. This will often present as Fanconi syndrome with multiple derangements of renal tubular reabsorption, including tubular acidosis with bicarbonate and phosphate wasting. These tubular abnormalities in GSD I are typically detected and monitored by urinary calcium. Long term these derangements can exacerbate uric acid nephropathy, otherwise driven by hyperlactatemia. In adolescence and beyond, glomerular disease may independently develop, initially presenting as glomerular hyperfiltration indicated by elevated urinary eGFR.
Splenomegaly
Enlargement of the spleen (splenomegaly) is common in GSD I and has two primary causes. In GSD Ia, splenomegaly may be caused by a relation between the liver and the spleen which causes either to grow or shrink to match the relative size of the other, to a lessened degree. In GSD Ib, it is a side effect of the use of filgrastim to treat neutropenia.
Bowel effects
Intestinal involvement can cause mild malabsorption with greasy stools (steatorrhea), but usually requires no treatment.
Infection risk
Neutropenia is a distinguishing feature of GSD Ib, absent in GSD Ia. The microbiological cause of neutropenia in GSD Ib is not well understood. Broadly, the problem arises from compromised cellular metabolism in the neutrophil, resulting in accelerated neutrophil apoptosis. The neutropenia in GSD is characterized by both a decrease in absolute neutrophil count and diminished neutrophil function. Neutrophils use a specific G6P metabolic pathway which relies on the presence of G6Pase-β or G6PT to maintain energy homeostasis within the cell. The absence of G6PT in GSD Ib limits this pathway, leading to endoplasmic reticulum stress, oxidative stress within the neutrophil, triggering premature apoptosis.[3] Granulocyte colony-stimulating factor (G-CSF), available as filgrastim, can reduce the risk of infection. In some cases, G-CSF formulated as pegfilgrastim, sold under the trade name Neulasta, may be used as a slow-acting alternative, requiring less frequent dosing.
Thrombocytopenia and blood clotting problems
Impaired platelet aggregation is an uncommon consequence of chronic hypoglycemia, seen in GSD I patients. Research has demonstrated decreased platelet function, characterized by decreased prothrombin consumption, abnormal aggregation reactions, prolonged bleeding time, and low platelet adhesiveness. Severity of platelet dysfunction typically correlates with clinical condition, with the most severe cases correlating with lactic acidosis and severely lipidemia.[25] It may cause clinically significant bleeding, especially epistaxis. Additionally, GSD I patients may present with thrombocytopenia as a consequence of splenomegaly. In the setting of splenomegaly various hematologic factors may be sequestered in the tissues of the spleen as blood is filtered through the organ. This can diminish levels of platelets available in the bloodstream, leading to thrombocytopenia.
Developmental effects
Developmental delay is a potential secondary effect of chronic or recurrent hypoglycemia, but is at least theoretically preventable. Normal neuronal and muscle cells do not express glucose-6-phosphatase, and are thus not impacted by GSD I directly. However, without proper treatment of hypoglycemia, growth failure commonly results from chronically low insulin levels, persistent acidosis, chronic elevation of catabolic hormones, and calorie insufficiency (or malabsorption). The most dramatic developmental delays are often the cause of severe (not just persistent) episodes of hypoglycemia.
Diagnosis
Several different problems may lead to the diagnosis, usually by two years of age:
- seizures or other manifestations of severe fasting hypoglycemia
- hepatomegaly with abdominal protuberance
- hyperventilation and apparent respiratory distress due to metabolic acidosis
- episodes of vomiting due to metabolic acidosis, often precipitated by minor illness and accompanied by hypoglycemia
Once the diagnosis is suspected, the multiplicity of clinical and laboratory features usually makes a strong circumstantial case. If hepatomegaly, fasting hypoglycemia, and poor growth are accompanied by lactic acidosis, hyperuricemia, hypertriglyceridemia, and enlarged kidneys by ultrasound, GSD I is the most likely diagnosis. The differential diagnosis list includes glycogenoses types III and VI, fructose 1,6-bisphosphatase deficiency, and a few other conditions (page 5), but none are likely to produce all of the features of GSD I.
The next step is usually a carefully monitored fast. Hypoglycemia often occurs within six hours. A critical blood specimen obtained at the time of hypoglycemia typically reveals a mild metabolic acidosis, high free fatty acids and beta-hydroxybutyrate, very low insulin levels, and high levels of glucagon, cortisol, and growth hormone. Administration of intramuscular or intravenous glucagon (0.25 to 1 mg, depending on age) or epinephrine produces little rise of blood sugar.
The diagnosis is definitively confirmed by liver biopsy with electron microscopy and assay of glucose-6-phosphatase activity in the tissue and/or specific gene testing, available in recent years.
Treatment
The primary treatment goal is prevention of hypoglycemia and the secondary metabolic derangements by frequent feedings of foods high in glucose or starch (which is readily digested to glucose). To compensate for the inability of the liver to provide sugar, the total amount of dietary carbohydrate should approximate the 24-hour glucose production rate. The diet should contain approximately 65–70% carbohydrate, 10–15% protein, and 20–25% fat. At least a third of the carbohydrates should be supplied through the night, so that a young child goes no more than 3–4 hours without carbohydrate intake. Once a diagnosis is made, the priority in GSD I treatment is to maintain an adequate blood glucose. Patients aim to maintain a blood glucose above the 72 mg/dL (4.0 mmol/L) cutoff for hypoglycemia. GSD Ib patients have an additional treatment priority relating to neutropenia. Proper management of blood glucose in GSD I is critical in avoiding the more severe effects of high levels of lactic acid and uric acid in the blood, and the development of hepatic adenomas.
In the last 30 years, two methods have been used to achieve this goal in young children: (1) continuous nocturnal gastric infusion of glucose or starch; and (2) night-time feedings of uncooked cornstarch. An elemental formula, glucose polymer, and/or cornstarch can be infused continuously through the night at a rate supplying 0.5–0.6 g/kg/h of glucose for an infant, or 0.3–0.4 for an older child. This method requires a nasogastric or gastrostomy tube and pump. Sudden death from hypoglycemia has occurred due to malfunction or disconnection, and periodic cornstarch feedings are now preferred to continuous infusion.
Cornstarch is an inexpensive way to provide gradually digested glucose. One tablespoon contains nearly 9 g carbohydrate (36 calories). Although it is safer, less expensive, and requires no equipment, this method does require that parents arise every 3–4 hours to administer the cornstarch. A typical requirement for a young child is 1.6 g/kg every 4 hours.
Long-term management should eliminate hypoglycemic symptoms and maintain normal growth. Treatment should achieve normal glucose, lactic acid, and electrolyte levels, and only mild elevations of uric acid and triglycerides.
Avoidance of other sugars
Intake of carbohydrates which must be converted to G6P to be utilized (e.g., galactose and fructose) should be minimized. Although elemental formulas are available for infants, many foods contain fructose or galactose in the forms of sucrose or lactose. Adherence becomes a contentious treatment issue after infancy.
Other therapeutic measures
Persistent elevation of uric acid above 6.5 mg/dl warrants treatment with allopurinol to prevent uric acid deposition in kidneys and joints.
Because of the potential for impaired platelet function, coagulation ability should be checked and the metabolic state normalized before surgery. Bleeding time may be normalized with 1–2 days of glucose loading, and improved with ddavp. During surgery, IV fluids should contain 10% dextrose and no lactate.
A patient with GSD, type 1b was treated with a liver transplant at UCSF Medical Center in 1993 that resulted in the resolution of hypoglycemic episodes and the need for the patient to stay away from natural sources of sugar. Other patients have undergone this procedure as well with positive results. Although a liver transplant resulted in the resolution of hypoglycemia it did not however resolve the chronic neutropenia and the risk of infection among patients.
Treatment of acute metabolic acidosis episodes
The most significant acute problem in childhood is a vulnerability to episodes of metabolic acidosis precipitated by minor illnesses. If a vomiting illness persists longer than 2–4 hours, the child should be seen and assessed for dehydration, acidosis, and hypoglycemia. If these are developing, intravenous fluids should be provided at a rate above maintenance. For mild acidosis, an effective fluid is 10% dextrose in ½ normal saline with 20 mEq/L KCl, but if acidosis is severe, 75–100 mEq/L NaHCO
3 and 20 mEq/L of K acetate can be substituted for the NaCl and KCl.
Metabolic control
Metabolic control often diminishes during and after puberty, as a result of a patient outgrowing their dietary treatment plan.[26]
Prognosis
Without adequate metabolic treatment, patients with GSD I have died in infancy or childhood of overwhelming hypoglycemia and acidosis. Those who survived were stunted in physical growth and delayed in puberty because of chronically low insulin levels. Intellectual disability resulting from recurrent, severe hypoglycemia is considered preventable with appropriate treatment.
Liver complications have been serious in some patients. Adenomas of the liver can develop in the second decade or later, with a small chance of later malignant transformation to hepatoma or hepatic carcinomas (detectable by alpha-fetoprotein screening). Several children with advanced hepatic complications have improved after liver transplantation.
Additional problems reported in adolescents and adults with GSD I have included hyperuricemic gout, pancreatitis, and chronic kidney failure. Despite hyperlipidemia, atherosclerotic complications are uncommon.
With diagnosis before serious harm occurs, prompt reversal of acidotic episodes, and appropriate long-term treatment, most children will be healthy. With exceptions and qualifications, adult health and life span may also be fairly good, although lack of effective treatment before the mid-1980s means information on long-term efficacy is limited.
Epidemiology
In the United States, GSD I has an incidence of approximately 1 in 50,000[27] to 100,000[28] births. None of the glycogenoses are currently detected by standard or extended newborn screening.
The disease is more common in people of Ashkenazi Jewish, Mexican, Chinese, and Japanese descent.[29]
References
- Veiga-da-Cunha, M.; Gerin, I.; Van Schaftingen, E. (May 2000). "How many forms of glycogen storage disease type I?". European Journal of Pediatrics. 159 (5): 314–318. doi:10.1007/s004310051279. ISSN 0340-6199. PMID 10834514.
- Kneeman, Jacob M.; Misdraji, Joseph; Corey, Kathleen E. (May 2012). "Secondary causes of nonalcoholic fatty liver disease". Therapeutic Advances in Gastroenterology. 5 (3): 199–207. doi:10.1177/1756283X11430859. ISSN 1756-283X. PMC 3342568. PMID 22570680.
- Chou, Janice Y.; Jun, Hyun Sik; Mansfield, Brian C. (January 2010). "Neutropenia in type Ib glycogen storage disease". Current Opinion in Hematology. 17 (1): 36–42. doi:10.1097/MOH.0b013e328331df85. ISSN 1065-6251. PMC 3099242. PMID 19741523.
- Dale, David C.; Bolyard, Audrey Anna; Marrero, Tracy M.; Phan, Lan; Boxer, Laurence A.; Kishnani, Priya S.; Kurtzberg, Joanne; Weinstein, David A. (2011-11-18). "Neutropenia in Glycogen Storage Disease 1b (GSD1b)". Blood. 118 (21): 4791. doi:10.1182/blood.V118.21.4791.4791. ISSN 0006-4971.
- Visser, Gepke; Rake, Jan Peter; Labrune, Philippe; Leonard, James V.; Moses, Shimon; Ullrich, Kurt; Wendel, Udo; Groenier, Klaas H.; Smit, G. Peter A. (October 2002). "Granulocyte colony-stimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1". European Journal of Pediatrics. 161 Suppl 1: S83–87. doi:10.1007/s00431-002-1010-0. ISSN 0340-6199. PMID 12373578. S2CID 33840498.
- "Glycogen Storage Disease Type I". NORD (National Organization for Rare Disorders). Retrieved 2019-09-29.
- Gierke's syndrome at Who Named It?
- von Gierke, E. (1929). "Hepato-nephromegalia glykogenica (Glykogenspeicherkrankheit der Leber und Nieren)". Beiträge zur Pathologischen Anatomie und zur Allgemeinen Pathologie. Jena. 82: 497–513.
- Parikh, Nirzar S.; Ahlawat, Rajni (2019), "Glycogen Storage Disease Type I (Von Gierke Disease)", StatPearls, StatPearls Publishing, PMID 30480935, retrieved 2019-11-01
- Jun, Hyun Sik; Weinstein, David A.; Lee, Young Mok; Mansfield, Brian C.; Chou, Janice Y. (2014-05-01). "Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib". Blood. 123 (18): 2843–2853. doi:10.1182/blood-2013-05-502435. ISSN 0006-4971. PMC 4007611. PMID 24565827.
- Kishnani, Priya S.; Austin, Stephanie L.; Abdenur, Jose E.; Arn, Pamela; Bali, Deeksha S.; Boney, Anne; Chung, Wendy K.; Dagli, Aditi I.; Dale, David; Koeberl, Dwight; Somers, Michael J. (November 2014). "Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics". Genetics in Medicine. 16 (11): e1. doi:10.1038/gim.2014.128. ISSN 1530-0366. PMID 25356975.
- Labrune, P.; Trioche, P.; Duvaltier, I.; Chevalier, P.; Odièvre, M. (March 1997). "Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature". Journal of Pediatric Gastroenterology and Nutrition. 24 (3): 276–279. doi:10.1097/00005176-199703000-00008. ISSN 0277-2116. PMID 9138172.
- Wang, David Q.; Carreras, Caroline T.; Fiske, Laurie M.; Austin, Stephanie; Boree, Danielle; Kishnani, Priya S.; Weinstein, David A. (September 2012). "Characterization and pathogenesis of anemia in glycogen storage disease type Ia and Ib". Genetics in Medicine. 14 (9): 795–799. doi:10.1038/gim.2012.41. ISSN 1098-3600. PMC 3808879. PMID 22678084.
- David, Weinstein (2019-09-04). "Ultragenyx DTX401 Phase 1/2 Cohort 2 Data Conference Call". edge.media-server.com. Retrieved 2019-10-30.
- Takamatsu, Yasushi; Jimi, Shiro; Sato, Tomohito; Hara, Shuuji; Suzumiya, Junji; Tamura, Kazuo (January 2007). "Thrombocytopenia in association with splenomegaly during granulocyte-colony-stimulating factor treatment in mice is not caused by hypersplenism and is resolved spontaneously". Transfusion. 47 (1): 41–49. doi:10.1111/j.1537-2995.2007.01061.x. ISSN 0041-1132. PMID 17207228. S2CID 36128181.
- Chen, Tzu-Lin; Chiang, Ya-Wen; Lin, Guan-Ling; Chang, Hsin-Hou; Lien, Te-Sheng; Sheh, Min-Hua; Sun, Der-Shan (2018-05-02). "Different effects of granulocyte colony-stimulating factor and erythropoietin on erythropoiesis". Stem Cell Research & Therapy. 9 (1): 119. doi:10.1186/s13287-018-0877-2. ISSN 1757-6512. PMC 5930863. PMID 29720275.
- "Glycogen storage disease type I". Genetics Home Reference from U.S. National Library of Medicine & National Institutes of Health. Retrieved 6 July 2013.
- Bali, DS; Chen, YT; Goldstein, JL; Pagon, RA; Adam, MP; Bird, TD; Dolan, CR; Fong, CT; et al. (1993). "Glycogen Storage Disease Type I". PMID 20301489.
{{cite journal}}: Cite journal requires|journal=(help) - Online Mendelian Inheritance in Man (OMIM): Glycogen Storage Disease Ib - 232220
- Online Mendelian Inheritance in Man (OMIM): Glycogen Storage Disease Ic - 232240
- PARKER, PAUL (1981). "Regression of hepatic adenomas in type Ia glycogen storage disease with dietary therapy". Gastroenterology. 81 (3): 534–536. doi:10.1016/0016-5085(81)90606-5. PMID 6941908.
- Minarich, Laurie A.; Kirpich, Alexander; Fiske, Laurie M.; Weinstein, David A. (2013). "Bone mineral density in glycogen storage disease type Ia and Ib". Genetics in Medicine. 14 (8): 737–741. doi:10.1038/gim.2012.36. ISSN 1098-3600. PMC 3884026. PMID 22481133.
- Soejima, K.; Landing, B. H.; Roe, T. F.; Swanson, V. L. (1985). "Pathologic studies of the osteoporosis of Von Gierke's disease (glycogenosis 1a)". Pediatric Pathology. 3 (2–4): 307–319. doi:10.3109/15513818509078791. ISSN 0277-0938. PMID 3867867.
- Pan, Bo-Lin; Loke, Song-Seng (2018-01-10). "Chronic kidney disease associated with decreased bone mineral density, uric acid and metabolic syndrome". PLOS ONE. 13 (1): e0190985. Bibcode:2018PLoSO..1390985P. doi:10.1371/journal.pone.0190985. ISSN 1932-6203. PMC 5761949. PMID 29320555.
- Czapek, Emily E.; Deykin, Daniel; Salzman, Edwin W. (1973-02-01). "Platelet Dysfunction in Glycogen Storage Disease Type I". Blood. 41 (2): 235–247. doi:10.1182/blood.V41.2.235.235. ISSN 0006-4971. PMID 4350560.
- Derks, Terry G. J.; van Rijn, Margreet (2015). "Lipids in hepatic glycogen storage diseases: pathophysiology, monitoring of dietary management and future directions". Journal of Inherited Metabolic Disease. 38 (3): 537–543. doi:10.1007/s10545-015-9811-2. ISSN 0141-8955. PMC 4432100. PMID 25633903.
- Glycogen-Storage Disease Type I at eMedicine
- https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/ Nation Organization for Rare Disorders
- Goldman, Lee (2011). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. pp. 1356. ISBN 978-1437727883.
Further reading
External links
 Media related to Glycogen storage disease type I at Wikimedia Commons
Media related to Glycogen storage disease type I at Wikimedia Commons