Ligand field theory
Ligand field theory (LFT) describes the bonding, orbital arrangement, and other characteristics of coordination complexes.[1][2][3] It represents an application of molecular orbital theory to transition metal complexes. A transition metal ion has nine valence atomic orbitals - consisting of five nd, one (n+1)s, and three (n+1)p orbitals. These orbitals are of appropriate energy to form bonding interaction with ligands. The LFT analysis is highly dependent on the geometry of the complex, but most explanations begin by describing octahedral complexes, where six ligands coordinate to the metal. Other complexes can be described by reference to crystal field theory.[4] Inverted ligand field theory (ILFT) elaborates on LFT by breaking assumptions made of relative metal and ligand orbital energies.
History
Ligand field theory resulted from combining the principles laid out in molecular orbital theory and crystal field theory, which describes the loss of degeneracy of metal d orbitals in transition metal complexes. John Stanley Griffith and Leslie Orgel[5] championed ligand field theory as a more accurate description of such complexes, although the theory originated in the 1930s with the work on magnetism of John Hasbrouck Van Vleck. Griffith and Orgel used the electrostatic principles established in crystal field theory to describe transition metal ions in solution and used molecular orbital theory to explain the differences in metal-ligand interactions, thereby explaining such observations as crystal field stabilization and visible spectra of transition metal complexes. In their paper, they proposed that the chief cause of color differences in transition metal complexes in solution is the incomplete d orbital subshells.[5] That is, the unoccupied d orbitals of transition metals participate in bonding, which influences the colors they absorb in solution. In ligand field theory, the various d orbitals are affected differently when surrounded by a field of neighboring ligands and are raised or lowered in energy based on the strength of their interaction with the ligands.[5]
Bonding
σ-bonding (sigma bonding)
In an octahedral complex, the molecular orbitals created by coordination can be seen as resulting from the donation of two electrons by each of six σ-donor ligands to the d-orbitals on the metal. In octahedral complexes, ligands approach along the x-, y- and z-axes, so their σ-symmetry orbitals form bonding and anti-bonding combinations with the dz2 and dx2−y2 orbitals. The dxy, dxz and dyz orbitals remain non-bonding orbitals. Some weak bonding (and anti-bonding) interactions with the s and p orbitals of the metal also occur, to make a total of 6 bonding (and 6 anti-bonding) molecular orbitals
.png.webp)
In molecular symmetry terms, the six lone-pair orbitals from the ligands (one from each ligand) form six symmetry adapted linear combinations (SALCs) of orbitals, also sometimes called ligand group orbitals (LGOs). The irreducible representations that these span are a1g, t1u and eg. The metal also has six valence orbitals that span these irreducible representations - the s orbital is labeled a1g, a set of three p-orbitals is labeled t1u, and the dz2 and dx2−y2 orbitals are labeled eg. The six σ-bonding molecular orbitals result from the combinations of ligand SALCs with metal orbitals of the same symmetry.
π-bonding (pi bonding)
π bonding in octahedral complexes occurs in two ways: via any ligand p-orbitals that are not being used in σ bonding, and via any π or π* molecular orbitals present on the ligand.
In the usual analysis, the p-orbitals of the metal are used for σ bonding (and have the wrong symmetry to overlap with the ligand p or π or π* orbitals anyway), so the π interactions take place with the appropriate metal d-orbitals, i.e. dxy, dxz and dyz. These are the orbitals that are non-bonding when only σ bonding takes place.
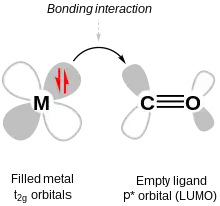
One important π bonding in coordination complexes is metal-to-ligand π bonding, also called π backbonding. It occurs when the LUMOs (lowest unoccupied molecular orbitals) of the ligand are anti-bonding π* orbitals. These orbitals are close in energy to the dxy, dxz and dyz orbitals, with which they combine to form bonding orbitals (i.e. orbitals of lower energy than the aforementioned set of d-orbitals). The corresponding anti-bonding orbitals are higher in energy than the anti-bonding orbitals from σ bonding so, after the new π bonding orbitals are filled with electrons from the metal d-orbitals, ΔO has increased and the bond between the ligand and the metal strengthens. The ligands end up with electrons in their π* molecular orbital, so the corresponding π bond within the ligand weakens.
The other form of coordination π bonding is ligand-to-metal bonding. This situation arises when the π-symmetry p or π orbitals on the ligands are filled. They combine with the dxy, dxz and dyz orbitals on the metal and donate electrons to the resulting π-symmetry bonding orbital between them and the metal. The metal-ligand bond is somewhat strengthened by this interaction, but the complementary anti-bonding molecular orbital from ligand-to-metal bonding is not higher in energy than the anti-bonding molecular orbital from the σ bonding. It is filled with electrons from the metal d-orbitals, however, becoming the HOMO (highest occupied molecular orbital) of the complex. For that reason, ΔO decreases when ligand-to-metal bonding occurs.
The greater stabilization that results from metal-to-ligand bonding is caused by the donation of negative charge away from the metal ion, towards the ligands. This allows the metal to accept the σ bonds more easily. The combination of ligand-to-metal σ-bonding and metal-to-ligand π-bonding is a synergic effect, as each enhances the other.
As each of the six ligands has two orbitals of π-symmetry, there are twelve in total. The symmetry adapted linear combinations of these fall into four triply degenerate irreducible representations, one of which is of t2g symmetry. The dxy, dxz and dyz orbitals on the metal also have this symmetry, and so the π-bonds formed between a central metal and six ligands also have it (as these π-bonds are just formed by the overlap of two sets of orbitals with t2g symmetry.)
High and low spin and the spectrochemical series
The six bonding molecular orbitals that are formed are "filled" with the electrons from the ligands, and electrons from the d-orbitals of the metal ion occupy the non-bonding and, in some cases, anti-bonding MOs. The energy difference between the latter two types of MOs is called ΔO (O stands for octahedral) and is determined by the nature of the π-interaction between the ligand orbitals with the d-orbitals on the central atom. As described above, π-donor ligands lead to a small ΔO and are called weak- or low-field ligands, whereas π-acceptor ligands lead to a large value of ΔO and are called strong- or high-field ligands. Ligands that are neither π-donor nor π-acceptor give a value of ΔO somewhere in-between.
The size of ΔO determines the electronic structure of the d4 - d7 ions. In complexes of metals with these d-electron configurations, the non-bonding and anti-bonding molecular orbitals can be filled in two ways: one in which as many electrons as possible are put in the non-bonding orbitals before filling the anti-bonding orbitals, and one in which as many unpaired electrons as possible are put in. The former case is called low-spin, while the latter is called high-spin. A small ΔO can be overcome by the energetic gain from not pairing the electrons, leading to high-spin. When ΔO is large, however, the spin-pairing energy becomes negligible by comparison and a low-spin state arises.
The spectrochemical series is an empirically-derived list of ligands ordered by the size of the splitting Δ that they produce. It can be seen that the low-field ligands are all π-donors (such as I−), the high field ligands are π-acceptors (such as CN− and CO), and ligands such as H2O and NH3, which are neither, are in the middle.
I− < Br− < S2− < SCN− < Cl− < NO3− < N3− < F− < OH− < C2O42− < H2O < NCS− < CH3CN < py (pyridine) < NH3 < en (ethylenediamine) < bipy (2,2'-bipyridine) < phen (1,10-phenanthroline) < NO2− < PPh3 < CN− < CO
See also
References
- Ballhausen, Carl Johan,"Introduction to Ligand Field Theory",McGraw-Hill Book Co., New York, 1962
- Griffith, J.S. (2009). The Theory of Transition-Metal Ions (re-issue ed.). Cambridge University Press. ISBN 978-0521115995.
- Schläfer, H. L.; Gliemann, G. "Basic Principles of Ligand Field Theory" Wiley Interscience: New York; 1969
- G. L. Miessler and D. A. Tarr "Inorganic Chemistry" 3rd Ed, Pearson/Prentice Hall, ISBN 0-13-035471-6.
- Griffith, J.S. and L.E. Orgel. "Ligand Field Theory". Q. Rev. Chem. Soc. 1957, 11, 381-393
External links
- Crystal-field Theory, Tight-binding Method, and Jahn-Teller Effect in E. Pavarini, E. Koch, F. Anders, and M. Jarrell (eds.): Correlated Electrons: From Models to Materials, Jülich 2012, ISBN 978-3-89336-796-2