Anti-neutrophil cytoplasmic antibody
Anti-neutrophil cytoplasmic antibodies (ANCAs) are a group of autoantibodies, mainly of the IgG type, against antigens in the cytoplasm of neutrophils (the most common type of white blood cell) and monocytes. They are detected as a blood test in a number of autoimmune disorders, but are particularly associated with systemic vasculitis, so called ANCA-associated vasculitides (AAV).

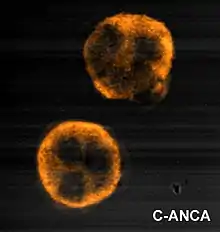
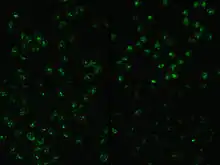
ANCA IF patterns
Immunofluorescence (IF) on ethanol-fixed neutrophils is used to detect ANCA, although formalin-fixed neutrophils may be used to help differentiate ANCA patterns. ANCA can be divided into four patterns when visualised by IF; cytoplasmic ANCA (c-ANCA), C-ANCA (atypical), perinuclear ANCA (p-ANCA) and atypical ANCA (a-ANCA), also known as x-ANCA. c-ANCA shows cytoplasmic granular fluorescence with central interlobular accentuation. C-ANCA (atypical) shows cytoplasmic staining that is usually uniform and has no interlobular accentuation. p-ANCA has three subtypes, classical p-ANCA, p-ANCA without nuclear extension and granulocyte specific-antinuclear antibody (GS-ANA). Classical p-ANCA shows perinuclear staining with nuclear extension, p-ANCA without nuclear extension has perinuclear staining without nuclear extension and GS-ANA shows nuclear staining on granulocytes only. a-ANCA often shows combinations of both cytoplasmic and perinuclear staining.[1]
ANCA antigens
The c-ANCA antigen is specifically proteinase 3 (PR3). p-ANCA antigens include myeloperoxidase (MPO) and bacterial permeability increasing factor Bactericidal/permeability-increasing protein (BPI). Other antigens exist for c-ANCA (atypical), however many are as yet unknown. Classical p-ANCA occurs with antibodies directed to MPO. p-ANCA without nuclear extension occurs with antibodies to BPI, cathepsin G, elastase, lactoferrin and lysozyme. GS-ANA are antibodies directed to granulocyte specific nuclear antigens. Atypical ANCA are thought to be antigens similar to that of the p-ANCAs, however may occur due to differences in neutrophil processing.[1]
Other less common antigens include HMG1 (p-ANCA pattern), HMG2 (p-ANCA pattern), alpha enolase (p and c-ANCA pattern), catalase (p and c-ANCA pattern), beta glucuronidase (p-ANCA pattern), azurocidin (p and c-ANCA pattern), actin (p and a-ANCA) and h-lamp-2 (c-ANCA).[1]
ELISA
Enzyme-linked immunosorbent assay (ELISA) is used in diagnostic laboratories to detect ANCAs. Although IF can be used to screen for many ANCAs, ELISA is used to detect antibodies to individual antigens. The most common antigens used on an ELISA microtitre plate are MPO and PR3, which are usually tested for after a positive IF test.[2]
Development
It is poorly understood how ANCA are developed, although several hypotheses have been suggested. There is probably a genetic contribution, particularly in genes controlling the level of immune response – although genetic susceptibility is likely to be linked to an environmental factor, some possible factors including vaccination or exposure to silicates. Two possible mechanisms of ANCA development are postulated, although neither of these theories answers the question of how the different ANCA specificities are developed, and there is much research still being undertaken on the development of ANCA.[3]
Theory of molecular mimicry
Microbial superantigens are molecules expressed by bacteria and other microorganisms that have the power to stimulate a strong immune response by activation of T-cells. These molecules generally have regions that resemble self-antigens that promote a residual autoimmune response – this is the theory of molecular mimicry. Staphylococcal and streptococcal superantigens have been characterized in autoimmune diseases – the classical example in post group A streptococcal rheumatic heart disease, where there is similarity between M proteins of Streptococcus pyogenes to cardiac myosin and laminin. It has also been shown that up to 70% of patients with granulomatosis with polyangiitis are chronic nasal carriers of Staphylococcus aureus, with carriers having an eight times increased risk of relapse.[3] This would therefore be considered a type II hypersensitivity reaction.
Theory of defective apoptosis
Neutrophil apoptosis, or programmed cell death, is vital in controlling the duration of the early inflammatory response, thus restricting damage to tissues by the neutrophils. ANCA may be developed either via ineffective apoptosis or ineffective removal of apoptotic cell fragments, leading to the exposure of the immune system to molecules normally sequestered inside the cells. This theory solves the paradox of how it could be possible for antibodies to be raised against the intracellular antigenic targets of ANCA.[3]
Role in disease
Disease associations
ANCAs are associated with small vessel vasculitides including granulomatosis with polyangiitis, microscopic polyangiitis, primary pauci-immune necrotizing crescentic glomerulonephritis (a type of renal-limited microscopic polyangiitis), eosinophilic granulomatosis with polyangiitis and drug induced vasculitides. PR3 directed c-ANCA is present in 80-90% of granulomatosis with polyangiitis, 20-40% of microscopic polyangiitis, 20-40% of pauci-immune crescentic glomerulonephritis and 35% of eosinophilic granulomatosis with polyangiitis. c-ANCA (atypical) is present in 80% of cystic fibrosis (with BPI as the target antigen) and also in inflammatory bowel disease, primary sclerosing cholangitis and rheumatoid arthritis (with antibodies to multiple antigenic targets). p-ANCA with MPO specificity is found in 50% of microscopic polyangiitis, 50% of primary pauci-immune necrotizing crescentic glomerulonephritis and 35% of eosinophilic granulomatosis with polyangiitis. p-ANCA with specificity to other antigens are associated with inflammatory bowel disease, rheumatoid arthritis, drug-induced vasculitis, autoimmune liver disease, drug induced syndromes and parasitic infections. Atypical ANCA is associated with drug-induced systemic vasculitis, inflammatory bowel disease and rheumatoid arthritis.[2][4] The ANCA‐positive rate is much higher in patients with type 1 diabetes mellitus than in healthy individuals.[5]
Levamisole, which is a common adulterant of cocaine, can cause an ANCA positive vasculitis.[6]
The presence or absence of ANCA cannot indicate presence or absence of disease and results are correlated with clinical features. The association of ANCA and disease activity remains controversial; however, the reappearance of ANCA after treatment can indicate a relapse.[7][8]
Pathogenesis
Although the pathogenic role of ANCA is still controversial, in vitro and animal models support the idea that the antibodies have a direct pathological role in the formation of small vessel vasculitides. MPO and PR3 specific ANCA can activate neutrophils and monocytes through their Fc and Fab'2 receptors, which can be enhanced by cytokines which cause neutrophils to display MPO and PR3 on their surface. Aberrant glycosylation of the MPO and PR3 specific ANCA enhances their ability to interact with activating Fc receptors on neutrophils.[9] The activated neutrophils can then adhere to endothelial cells where degranulation occurs. This releases free oxygen radicals and lytic enzymes, resulting in damage to the endothelium via the induction of necrosis and apoptosis. Furthermore, neutrophils release chemoattractive signalling molecules that recruit more neutrophils to the endothelium, acting as a positive feedback loop. Animal models have shown that MPO antibodies can induce necrotizing crescentic glomerulonephritis and systemic small vessel vasculitis. In these animal models the formation of glomerulonephritis and vasculitis can occur in the absence of T-cells, however neutrophils must be present.[10][11][12][13] Although ANCA titres have been noted to have limited correlation with disease activity, except for kidney disease, and with risk of relapse, this is explained by differences in the epitopes and affinity of ANCAs.[14] ANCAs induce excess activation of neutrophils, resulting in the production of neutrophil extracellular traps (NETs), which cause damage to small blood vessels.[14] In addition, in patients with active disease, treated with Rituximab, an anti-CD20 antibody which remove circulating B-cells, clinical remission correlates more to the decreasing number of circulating B-cells than decrease in ANCA titre, which in some patient does not change during treatment. The same study found that clinical relapse in some patients were in association with the return of circulating B-cells.[15] Based on the above observations and that ANCA reactive B-cells can be found in circulation in patients with AAV, an alternative hypothesis have been proposed assigning a direct pathogenic role of these cells, whereby activated neutrophils and ANCA-reactive B-cells engage in intercellular cross-talk, which leads not only to neutrophil degranulation and inflammation but also to the proliferation and differentiation of ANCA-reactive B-cells.[16] However, this hypothesis remains to be tested.
History
ANCAs were originally described in Davies et al. in 1982 in segmental necrotising glomerulonephritis.[14][18] The Second International ANCA Workshop, held in The Netherlands in May 1989, fixed the nomenclature on perinuclear vs. cytoplasmic patterns, and the antigens MPO and PR3 were discovered in 1988 and 1989, respectively.[19] International ANCA Workshops have been carried out every two years.
References
- Mead, A.R. Bradwell, R.P. Stokes, G.P. (1999). Advanced atlas of autoantibody patterns. Birmingham: The Binding Site. ISBN 978-0704485105.
- Savige, J; Davies, D; Falk, RJ; Jennette, JC; Wiik, A (Mar 2000). "Antineutrophil cytoplasmic antibodies and associated diseases: a review of the clinical and laboratory features". Kidney International. 57 (3): 846–62. doi:10.1046/j.1523-1755.2000.057003846.x. PMID 10720938. S2CID 14521707.
- Reumaux D, Duthilleul P, Roos D (2004). "Pathogenesis of diseases associated with antineutrophil cytoplasm autoantibodies". Hum Immunol. 65 (1): 1–12. doi:10.1016/j.humimm.2003.09.013. PMID 14700590.
- Bossuyt, X (February 2006). "Serologic markers in inflammatory bowel disease". Clinical Chemistry. 52 (2): 171–81. doi:10.1373/clinchem.2005.058560. PMID 16339302.
- Omura, T (September 2019). "Oldest‐old type 1 diabetes patient receiving insulin pump treatment with positive myeloperoxidase‐antineutrophil cytoplasmic antibody complication: A case report". Geriatrics & Gerontology International. 19 (9): 957–58. doi:10.1111/ggi.13683. PMID 31490005. S2CID 201845957.
- Tran, H; Tan, D; Marnejon, TP (February 2013). "Cutaneous vasculopathy associated with levamisole-adulterated cocaine". Clinical Medicine & Research. 11 (1): 26–30. doi:10.3121/cmr.2012.1085. PMC 3573092. PMID 22723468.
- Sinclair, D; Stevens, JM (Sep 2007). "Role of antineutrophil cytoplasmic antibodies and glomerular basement membrane antibodies in the diagnosis and monitoring of systemic vasculitides". Annals of Clinical Biochemistry. 44 (Pt 5): 432–42. doi:10.1258/000456307781646049. PMID 17761028.
- Stegeman, CA (Nov 2005). "Predictive value of antineutrophil cytoplasmic antibodies in small-vessel vasculitis: is the glass half full or half empty?". The Journal of Rheumatology. 32 (11): 2075–7. PMID 16265681.
- Maverakis E, Kim K, Shimoda M, Gershwin M, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB (2015). "Glycans in the immune system and The Altered Glycan Theory of Autoimmunity". J Autoimmun. 57 (6): 1–13. doi:10.1016/j.jaut.2014.12.002. PMC 4340844. PMID 25578468.
- Falk, RJ; Jennette, JC (May 2010). "ANCA disease: where is this field heading?". Journal of the American Society of Nephrology. 21 (5): 745–52. doi:10.1681/ASN.2009121238. PMID 20395376.
- Seo, P; Stone, JH (Jul 1, 2004). "The antineutrophil cytoplasmic antibody-associated vasculitides". The American Journal of Medicine. 117 (1): 39–50. doi:10.1016/j.amjmed.2004.02.030. PMID 15210387.
- Jennette, JC; Xiao, H; Falk, RJ (May 2006). "Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies". Journal of the American Society of Nephrology. 17 (5): 1235–42. doi:10.1681/ASN.2005101048. PMID 16624929.
- Falk RJ, Terrell RS, Charles LA, Jennette JC. (1990). "Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro". Proc Natl Acad Sci U S A. 87 (11): 4115–4119. Bibcode:1990PNAS...87.4115F. doi:10.1073/pnas.87.11.4115. PMC 54058. PMID 2161532.
{{cite journal}}: CS1 maint: uses authors parameter (link) - Nakazawa, D; Masuda, S; Tomaru, U; Ishizu, A (February 2019). "Pathogenesis and therapeutic interventions for ANCA-associated vasculitis" (PDF). Nature Reviews Rheumatology. 15 (2): 91–101. doi:10.1038/s41584-018-0145-y. hdl:2115/74654. PMID 30542206. S2CID 54474335.
- Jayne DR, Jones RB; Ferraro AJ; Chaudhry AN; Brogan P; Salama AD; Smith KG; Savage CO (2009-07-01). "A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis". Arthritis Rheum. 60 (7): 2156–68. doi:10.1002/art.24637. PMID 19565480.
- Hurtado, Plinio; Nitschke, J.; Hurtado-Perez, E.; Peh, C.A. (April 2013). "ANCA reactive B cells and neutrophils cross-talk in the pathogenesis of AAV: A model proposal". La Presse Médicale. 42 (n° 4P2): 720. doi:10.1016/j.lpm.2013.02.256.
- "ChemoCentryx Announces FDA Approval of Tavneos (avacopan) in ANCA-Associated Vasculitis". ChemoCentryx, Inc. (Press release). 8 October 2021. Retrieved 11 October 2021.
- Davies, DJ; Moran, JE; Niall, JF; Ryan, GB (Aug 28 – Sep 4, 1982). "Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology?". British Medical Journal (Clinical Research Ed.). 285 (6342): 606. doi:10.1136/bmj.285.6342.606. PMC 1499415. PMID 6297657.
- Jennette, JC; Hoidal, JR; Falk, RJ (Jun 1, 1990). "Specificity of anti-neutrophil cytoplasmic autoantibodies for proteinase 3". Blood. 75 (11): 2263–4. doi:10.1182/blood.V75.11.2263.2263. PMID 2189509.
External links
- images of pANCA and cANCA
- fluorescence images of ANCA
- Anti-Neutrophil+Cytoplasmic+Antibody at the US National Library of Medicine Medical Subject Headings (MeSH)