Acecainide
Acecainide (N-acetylprocainamide, NAPA) is an antiarrhythmic drug. Chemically, it is the N-acetylated metabolite of procainamide. It is a Class III antiarrhythmic agent, whereas procainamide is a Class Ia antiarrhythmic drug. It is only partially as active as procainamide; when checking levels, both must be included in the final calculation.
 | |
| Clinical data | |
|---|---|
| Routes of administration | Oral or intravenous administration IV |
| Pharmacokinetic data | |
| Bioavailability | Oral administration 85% [1] |
| Protein binding | 10% [2] |
| Metabolism | Hepatic |
| Elimination half-life | 4.3–15.1 h [2] |
| Excretion | Renal |
| Identifiers | |
IUPAC name
| |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.151.497 |
| Chemical and physical data | |
| Formula | C15H23N3O2 |
| Molar mass | 277.368 g·mol−1 |
| 3D model (JSmol) | |
| Density | 1.1±0.1 g/cm3 g/cm3 |
| Melting point | 138–140 °C (280–284 °F) |
| Boiling point | 465–535 °C (869–995 °F) |
SMILES
| |
History
In the early 1930s, Claude Beck was undertaking pioneer cardiac surgery at the Lakeside Hospital in Cleveland, Ohio. During and after his surgery he was facing problems with arrhythmias. These problems were investigated by Frederick R. Mautz. In these experiments he used drugs similar to cocaine, because these drugs were readily absorbed from mucous membranes and were also known to have some effect on the myocardium. Mautz tried procaine, but its action was short-lived owing to digestion by esterases. From procaine Mautz synthesized procainamide, which is not a substrate for esterases. Procainamide has the additional advantage of being active by mouth. Procainamide was approved by the US FDA on June 2, 1950 under the brand name Pronestyl. In 1951 Pronestyl was launched by Bristol-Myers Squibb, a pharmaceutical company in the US. Along with the discovery of procainamide came the discovery of its metabolite acecainide.[3]
Synthesis
Procainamide is metabolized in the liver to acecainide by N-acetyltransferase, an enzyme that is genetically determined.[4] N-Acetyltransferase is an enzyme that catalyzes the transfer of acetyl groups from acetyl-CoA to arylamines and aromatic amines such as procainamide.[5]
This reaction is known as an acetylation reaction, that refers to the process of introducing an acetyl group (resulting in an acetoxy group) into a compound, namely the substitution of an acetyl group for an active hydrogen atom.
Activity
Acecainide is the major metabolite of the antiarrhythmic drug, procainamide. Measurements of procainamide in serum may not accurately reflect the drug's complete pharmacologic activity in the body. Monitoring acecainide levels along with the procainamide is recommended during procainamide therapy. Acecainide is considered comparable in activity to its parent compound; however, acecainide levels will vary widely. Serum levels of acecainide increase in patients on chronic procainamide therapy, particularly in those with renal impairment. The average serum concentration ratio of acecainide to procainamide is 0.8 to 1.2, depending on a genetically determined tendency to acetylate procainamide rapidly or slowly. Because the ratio varies from patient to patient, measuring serum acecainide and procainamide together helps to achieve an optimum anti-arrhythmic effect and reduce the risk of toxicity.[6]
Mechanism
Acecainide is a potassium-channel blocker like Class III antiarrhythmic compounds. This compounds can bind to potassium channels and delays phase 3 repolarization. These electrophysiological changes decrease the sensitiveness of cells to electrical stimuli, which leads to an increase in action potential duration and an increase in the effective refractory period. By increasing the effective refractory period NAPA is very useful in suppressing tachyarrhythmias caused by re-entry ventricular arrhythmia.[7] In this way NAPA can increase the Q - T interval of the PQRST heart rhythm.[8]

Medical uses
Acecainide is pharmacologically active as an antiarrhythmic agent. It has electrophysiological effects of a class III antiarrhythmic drug and it is used as a medicine to increase the Q – T interval of the PQRST heart rhythm in patients with cardiac arrhythmias. The equivalent drug procainamide, which is a class Ia antiarrhythmic drug, is also used in patients with cardiac arrhythmias.[8] Nevertheless, NAPA does only affect the Q - T interval, while procainamide has also effect on the QRS-interval.[9] Also the electrophysiologic properties of NAPA are a little bit different from those of procainamide and NAPA is not completely effective for suppressing ventricular dysrhythmias, but its antiarrhythmic mechanisms are similar to those of procainamide.[10]
Toxicokinetics
Absorption and plasma concentrations
The pharmacokinetic properties of acecainide, an active metabolite of procainamide, have been studied in healthy people and patients with cardiomyopathy in elderly and younger patients. In healthy people, mean peak plasma concentrations following oral doses of 900 and 1000 mg of NAPA were 5.9 and 5.3 mg/L, and it attained 2.2 to 2.8 hours after administration. In a few patients with cardiomyopathy the mean peak plasma concentration was 5.6 mg/L 1.6 hours after ingestion. Acecainide has a bioavailability of 85%, with a mean peak plasma occurring between 45 and 90 minutes.[11]
Distribution
The mean apparent volume of distribution range is from 2.61 to 2.9 L/kg in healthy people and patients with cardiomyopathy. At steady state, the volume of distribution is 1.3 to 1.7 L/kg in healthy subjects, 1.3 to 1.58 L/kg in patients with coronary artery disease, and 1.25 L/kg in patients with ventricular arrhythmias receiving acecainide therapy.[11]
Acecainide has a volume of distribution of 1.5 L/kg that is less than the Vd of procainamide (2.0 L/kg). Also it binds 10% less to proteins than procainamide. Because of this low volume of distribution, it can be concluded that the medicine is thought to be confined in the plasma or liquid parts of the blood.[12][13]
Metabolism and elimination
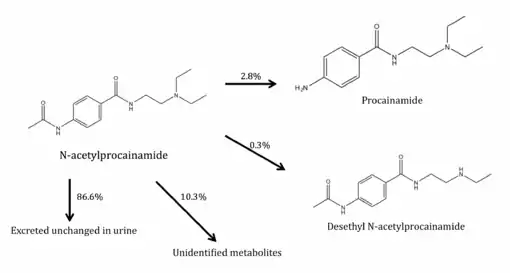
In the body, acecainide is metabolised in several products. Some acecainide can convert to procainamide. The deacetylation clearance of acecainide is 0.39 L/h compare to a total NAPA clearance of 1.38 L/h, indicating that 2.8% of NAPA was converted to procainamide, 0.3% was desethylated, and 10.3% convert to unidentified metabolites, with 86.6% excreted unchanged. Following oral administration, 59 to 87% of a dose of acecainide is excreted unchanged in the urine.[11]
Renal clearance of acecainide following short and long term administration ranges from 2.08±0.36 mL/min/kg to 3.28±0.52 mL/min/kg in healthy people. There is a linear relationship between acecainide clearance and creatinine clearance.[2]
However, the clearance of acecainide was reduced in a few patients with cardiomyopathyand and ventricular arrhythmias. Also is excretion of procainamide and NAPA is reduced in patients with CKD.[14] A major reaction to procainamide and several other drugs is a syndrome that closely resembles lupus. A reactive metabolite, possibly nitrosoprocainamide is thought to play a role in the lupus reaction. Acecainide, unlike procainamide, appears not to form a reactive metabolite.
Mean plasma elimination half-lives in healthy subjects range from 6.8 to 9.6 hours following single or repeated doses when sampling times of 12 to 24 hours are used. A tendency for prolongation of half-lives was noted in patients with cardiomyopathy and in patients with arrhythmias.[11]
Influence on clearance and concentration rate
Steady-state procainamide and acecainide concentrations were used to compute procainamide clearance and acecainide/procainamide (NAPA/PA) concentration ratio. Using stepwise multiple linear regression age, creatinine clearance and congestive heart failure were found to influence procainamide clearance significantly (p less than 0.05). Age and creatinine clearance effected the NAPA/PA concentration ratio (p less than 0.05). Based on this data, age appears to have an independent effect on both procainamide clearance and the NAPA/PA ratio that is separate from the decline in renal function that occurs in elderly patients.[15]
Administration
It can be given either intravenously or orally, and is eliminated primarily by renal excretion. Comparative studies with other antiarrhythmic drugs have not been undertaken apart from a small study in atrial flutter where NAPA was better than quinidine plus digoxin. Although further clinical experience is required before the relative place of acecainide in therapy can be determined, the drug nevertheless appears to offer advantages over procainamide, particularly with respect to the reduced formation of antinuclear antibodies.
The dose of acecainide should be adjusted to control the patients’ arrhythmias and with regard to their clinical state including age, renal function and concurrent administration of other drugs. There appears to be an overlap in plasma concentrations required for therapeutic efficacy and those associated with adverse effects. Rapid bolus infusion of acecainide has been associated with serious hypotension and a maximum infusion rate of 50 mg/min has been suggested.[11]
Intravenous administration
Intravenous infusion of 0.45 mg/kg/min for 30 minutes, followed by 0.22 mg/mL/min for 30 minutes and a maintenance infusion, suppressed more than 90% of PVCs. Doses of acecainide 15 to 20 mg/kg were also effective in preventing induced ventricular tachycardia or prolonging ventricular tachycardia cycle length and reducing frequency of PVCs. However, the dosage required to maintain antiarrhythmic effects is still not clear.[11]
Oral administration
In short-term and more prolonged studies, acecainide 1.5 to 2.0 g as a single dose or as three or four divided doses a day appears to be satisfactory in controlling PVCs. In patients who are refractory to other antiarrhythmic agents, the doses required appear to be greater. Moreover, a few studies inferred that tolerance may develop with long-term administration of NAPA, necessitating increased doses. There are no specific dosage recommendations for patients with left ventricular dysfunction, although care should be taken to reduce the dose in patients with moderate to severe renal dysfunction.[11]
The loading dose of this medicine is 15–18 mg/kg administered over 30 to 60 minutes for people without insufficient renal function or decreased cardiac output and 12 mg/kg for people with this disorders. This can also be described by the formula of the loading dose:
LD = Vss(l/kg) × IBW(kg) × Cp(mg/L) / ( S x F)
The daily empiric oral dose can be calculated by the next formula:
Daily dose = (Css)ave × Cl × 1440 / ( S x F )
This those is decreased by 25% in patients with heart and/or liver failures, and it is decreased by 50% by patients with renal failures.[11]
Drug interaction
Research shows that accumulation of acecainide during procainamide therapy can alter both procainamide elimination as well as its electrophysiologic actions.[9] A few healthy people demonstrated that concomitantly administered trimethoprim decreased the renal clearance of procainamide and formed NAPA, resulting in increased plasma concentrations of both drugs and increases in QTc after procainamide. Trimethoprim, procainamide and acecainide are all excreted by active tubular secretion. Also amiodarone, cimetidine and trimethoprim increase the NAPA serum level. Cimetidine and ranitidine possibly increase plasma procainamide and NAPA concentrations and subsequent toxicity.[11] Furthermore, alcohol enhances acetylation of procainamide to NAPA and alcohol consumption may reduce half-life.[16] It also has been shown that coadministration of para-aminobenzoic acid decreases the biotransformation of procainamide to acecainide in a patient with rapid acetylation kinetics.[4]
Toxicity
Measuring the ratio of acecainide and procainamide concentrations together helps to achieve an optimum anti-arrhythmic effect and reduce the risk of toxicity.[6]
Effects on humans
acecainide can cause cardiac toxicity that effects in torsades de pointes. Also acecainide can decrease renal function when it is accumulated during a procainamide therapy. Furthermore, acecainide can lead to a low blood pressure and a serious left ventricle depression when it is present in toxic concentrations.[12] Other common side effects of NAPA are gastrointestinal disturbances, insomnia, dizziness, lightheadedness, blurred vision, numbness and tingling sensation.[13][17] There has been a large resemblance reported between the concentration range associated with arrhythmic suppression of acecainide and the range of concentrations where intolerable side effects begin to develop. No severe cardiac toxicity has been notified with oral intake despite plasma concentrations as high as 40 micrograms/mL. However, hypotension has been reported in association with a rapid injection of acecainide.[2]
Effects on animals
In animals, acecainide has positive inotropic effects, but it also causes negative chronotropic and hypotensive activity.[12] The cardiovascular pharmacodynamics of procainamide and NAPA have not been well identified in small rodents without the presence of anesthesia or restraint. Researchers are trying to make models of the effects of procainamide and acecainide in animals.[18]
Detoxification
1-Aminobenzotriazole (ABT) is a nonselective inhibitor of cytochrome P450 enzymes, but recent research confirmed that this inhibitor also inhibit the RLS9-catalyzed N-acetylation of procainamide. In rats oral ABT decreases the clearance of intravenous procainamide for 45%, followed by a decreased acecainide-to-procainamide ratio in urine and plasma. Studies in humans also shows that ABT is an inhibitor of N-acetyltransfersase. These studies claim that ABT is a more potent inhibitor of N-acetyltransferase 2 compared with N-acetyltransferase 1.[19]
References
- Strong JM. Dutcher JS. (1975). "Absolute bioavailability in man of N-acetylprocainamide determined by a novel stable isotope method". Clin Pharmacol Ther. 18 (5 Pt 1): 613–22. doi:10.1053/j.ajkd.2013.02.358. PMC 3851309. PMID 1183141.
- Connolly, S. J.; Kates, R. E. (1982). "Clinical pharmacokinetics of N-acetylprocainamide". Clinical Pharmacokinetics. 7 (3): 206–20. doi:10.2165/00003088-198207030-00002. PMID 6178545.
- Hollman A (February 1992). "Procaine and procainamide". Br Heart J. 67 (2): 143. doi:10.1136/hrt.67.2.143. PMC 1024743. PMID 18610401.
- Nylen, E. S.; Cohen, A. I.; Wish, M. H.; Lima, J. J.; Finkelstein, J. D. (1986). "Reduced acetylation of procainamide by para-aminobenzoic acid". Journal of the American College of Cardiology. 7 (1): 185–7. doi:10.1016/s0735-1097(86)80280-7. PMID 3484486.
- Evans DA (1989). "N-acetyltransferase". Pharmacology & Therapeutics. 42 (2): 157–234. doi:10.1016/0163-7258(89)90036-3. PMID 2664821.
- Mulberg E, Dalton P, Hefner A (1992). "N-Acetylprocainamide Assay". Clin Chem. 38 (6): 336.
- Cardiovascular physiology concepts. Lippincott Williams & Wilkins. 2011. p. 235. ISBN 9781451113846.
- Schoenwald, Ronald D., ed. (2002). Pharmacokinetics in drug discovery and development. CRC Press. ISBN 9781566769730.
- Funck-Brentano C (1989). "Pharmacokinetic and pharmacodynamic interaction of N-acetyl procainamide and procainamide in humans". J Cardiovasc Pharmacol. 14 (3): 364–73. doi:10.1097/00005344-198909000-00003. PMID 2476614. S2CID 27698759.
- Sung, R. J.; Juma, Z; Saksena, S (1983). "Electrophysiologic properties and antiarrhythmic mechanisms of intravenous N-acetylprocainamide in patients with ventricular dysrhythmias". American Heart Journal. 105 (5): 811–9. doi:10.1016/0002-8703(83)90245-4. PMID 6189384.
- Harron DW, Brogden RN (1990). "Acecainide (N-acetylprocainamide). A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in cardiac arrhythmias". Drugs. 39 (5): 720–40. doi:10.2165/00003495-199039050-00007. PMID 1693889. S2CID 46978066.
- Descotes, Jacques, ed. (1996). Human Toxicology. Elsevier. ISBN 978-0-444-81557-6.
- HCRC DESK Reference of clinical pharmacology. Elsevier. 1997. ISBN 9780849396830.
- Mohamed, Ahmed N.; et al. (2013). "Pharmacokinetic modeling and simulation of procainamide and N-acetylprocainamide in a patient receiving continuous renal replacement therapy: a novel approach to guide renal dose adjustments". American Journal of Kidney Diseases. 61 (6): 1046–1048. doi:10.1053/j.ajkd.2013.02.358. PMC 3851309. PMID 23562328.
- Bauer LA, Black D, Gensler A (1989). "Influence of age, renal function, and heart failure on procainamide clearance and n-acetylprocainamide serum concentrations". Int J Clin Pharmacol There Tox. 27 (5): 213–216. PMID 2472362.
- Olsen, H; Mørland, J (1982). "Ethanol-induced increase in procainamide acetylation in man". British Journal of Clinical Pharmacology. 13 (2): 203–8. doi:10.1111/j.1365-2125.1982.tb01357.x. PMC 1402013. PMID 7059417.
- Atkinson Jr, A. J.; Lee, W. K.; Quinn, M. L.; Kushner, W; Nevin, M. J.; Strong, J. M. (1977). "Dose-ranging trial of N-acetylprocainamide in patients with premature ventricular contractions". Clinical Pharmacology and Therapeutics. 21 (5): 575–87. doi:10.1002/cpt1977215575. PMID 322922. S2CID 22856741.
- Kharidia, J; Eddington, N. D. (1996). "Application of computer-assisted radiotelemetry in the pharmacokinetic and pharmacodynamic modeling of procainamide and N-acetylprocainamide". Journal of Pharmaceutical Sciences. 85 (6): 595–9. doi:10.1021/js950473h. PMID 8773955.
- Sun, Q; Harper, T. W.; Dierks, E. A.; Zhang, L; Chang, S; Rodrigues, A. D.; Marathe, P (2011). "1-Aminobenzotriazole, a known cytochrome P450 inhibitor, is a substrate and inhibitor of N-acetyltransferase". Drug Metabolism and Disposition. 39 (9): 1674–9. doi:10.1124/dmd.111.039834. PMID 21677062. S2CID 20848730.