Actin remodeling of neurons
Actin remodeling is a biochemical process in cells. In the actin remodeling of neurons, the protein actin is part of the process to change the shape and structure of dendritic spines. G-actin is the monomer form of actin, and is uniformly distributed throughout the axon and the dendrite. F-actin is the polymer form of actin, and its presence in dendritic spines is associated with their change in shape and structure. Actin plays a role in the formation of new spines as well as stabilizing spine volume increase.[1] The changes that actin brings about lead to the formation of new synapses as well as increased cell communication.
Actin remodeling consists of the dynamic changes in actin polymerization that underlie the morphological changes at the neural synapse. Actin is only able to cause all of the changes that promote long-term potentiation (LTP) through its formation from G-actin into F-actin. When F-actin is unable to form, long-term depression (LTD) is induced, which promotes opposite results. Stimulation of the neuron that promotes LTP causes larger spine volume, increased cell communication, and a greater ratio of F-actin to G-actin. In the LTD environment, spine volume is decreased, cell communication is decreased, and there is a far greater ratio of G-actin to F-actin.
Structural overview of actin
Actin exists in two states in the axonal and dendritic processes: globular or G-actin and filament/filamentous or F-actin. G-actin are the monomer building blocks that assemble via weak noncovalent interactions to form F-actin. F-actin is a two-stranded asymmetrical helical polymer. The asymmetrical quality of F-actin allows for different binding specificities at each end. One end shows an indentation and is referred to as the barbed end while the other resembles an arrow head and is referred to as the pointed end.
F-actin can be found in the presynaptic bouton surrounding synaptic vesicle clusters and acting as scaffolding.[1] Additionally, actin is present at the active zone and plays a role in moving vesicles to the active zone for exocytosis into the synapse. The active zone is the portion of the presynaptic membrane opposite the postsynaptic density across the synaptic cleft. It is the site of synaptic vesicle docking and neurotransmitter release.[2] Postsynaptically, F-actin can be found in the postsynaptic density zone (PSDZ) and throughout the spine head and neck. G-actin is uniformly distributed throughout the axon and the dendrite.[1]
The balance of F and G-actin is in a constant state of flux, which can be attributed to actin treadmilling. Actin treadmilling is the process of turnover of actin filaments where F-actin is rapidly assembled and disassembled. G-actin subunits preferentially add to the barbed end of the F-actin polymer and older units are removed from the pointed end. The concentration of free G-actin monomers decreases until it reaches a critical concentration where the rate of assembly to disassembly or the F to G-actin ratio reaches a steady state.
Role in short-term synaptic communication
Non-LTP inducing stimuli cause alterations in spine morphology due to changes in actin polymerization. Presynaptically, axonal boutons undergo submicron displacements that indent the dendritic spines.[3] Postsynaptically, innervation causes dendritic spines to remodel by as much as 30% over a period of seconds.[4] The spines show a lateral expansion that envelops the presynaptic axonal indentation. The changes due to non-LTP inducing stimuli dissipate after 5 minutes.[3]
Role in LTP and LTD
Actin is necessary for the induction of LTP. This protein allows for many changes both presynaptically and postsynaptically.
In the presynaptic region, actin allows for the formation of new axonal branches that result in new boutons. It also facilitates vesicle recruitment to the bouton.
Postsynaptically, actin filaments traffic AMPA receptors to the PSDZ, while also providing scaffolding for plasticity products such as CAMKII.[5] F-actin could serve as a synaptic tag because the scaffolding space for plasticity products is increased during LTP actin polymerization. Furthermore, the actin cytoskeleton in the neck of the spine compartmentalizes the LTP induced response to the innervated dendritic spine, which leads to the specificity of LTP.[6] Actin plays a role in the formation of new spines as well as stabilized spine volume increase.[1] All of these changes that actin brings about leads to the formation of new synapses as well as increased cell communication.
LTP inducing high frequency stimulation leads to NMDA receptor activation and calcium influx. Rho GTPases are then activated to polymerize G-actin to F-actin through the activity of actin binding proteins. An increase in the F-actin/G-actin ratio is observed 40 seconds after the LTP inducing stimulus.[7] The increase in polymerized F-actin is due to the recruitment of G-actin monomers and the translation of actin mRNA in the dendrite.[8] The stimulus induced change persists for approximately 5 weeks.[9]
Actin is only able to cause changes that promote LTP through its formation into F-actin. When F-actin is unable to form, LTD is induced, which promotes opposite results.
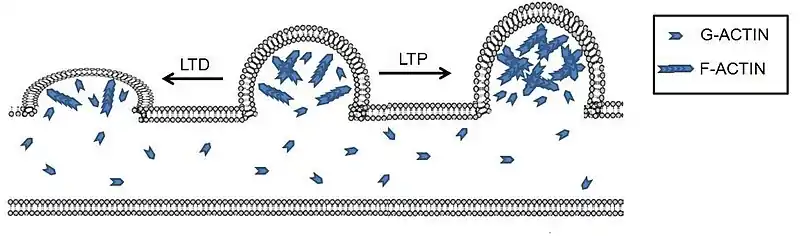
This figure demonstrates the morphological effects on the dendrites in LTP and LTD environments. In LTP we can see the larger spine volume as well as a greater ratio of F-actin to G-actin. This demonstrates the role of actin in LTP as well as the increased communication LTP creates. In the LTD environment, spine volume is decreased and there is a far greater ratio of G-actin to F-actin, demonstrating the importance of the F-actin to G-actin ratios in both LTP and LTD.
Actin binding proteins in LTP and LTD
Actin binding proteins prove significant in actin remodeling, as the LIMK1/ADF/Cofilin Pathway facilitates the development of F-actin. Actin Depolymerizing Factor, or ADF, normally disassembles actin and hampers the induction of LTP. However, synaptic activity favors the activation of LIMK1, a protein that phosphorylates the ADF/Cofilin complex at its phosphorylation site, Ser3, which inactivates the complex, promoting the formation of F-actin. If this pathway is disrupted, then G-actin is unable to polymerize and LTP is inhibited. One particular actin binding protein that plays a major role in disrupting this pathway is Gelsolin.[9] This protein caps the barbed end of F-actin, thus preventing G-actin subunits from binding to F-actin and blocking actin treadmilling. Activation of Gelsolin not only blocks LTP, but induces LTD. In LTD, the F to G-actin ratio is shifted towards G-actin and leads to a decrease in spine volume, as well as the occasional disappearance of spines altogether.
Implications for learning and memory
Being associated with long term structural changes at the synapse and LTP, it is no surprise that actin dynamics influence learning and memory. Experiments have shown that drugs like cytochalasin C and Latrunculin A that inhibit the assembly of G-actin into F-actin disrupt both the acquisition and extinction of fear responses in mice.[10] Disruption of actin dynamics can also affect visuospatial learning.[6]
LIMK1, an actin binding protein, phosphorylates ADF/cofilin, allowing the formation of F-actin.[9] LIMK1 knockout neurons are unable to form a cytoskeletal matrix within the dendritic spine,[6] which has interesting implications for learning. One of the primary functions of actin is to compartmentalize a neuron's response to stimulation – that is, to keep molecules essential for LTP within the stimulated spine.[6] Upon low frequency stimulation of knockout cells, these molecules are likely to diffuse out of the cell before a concentration significant enough to produce LTP builds up. Upon high frequency stimulation, however, there is an overabundance of these essential molecules, which are present in high enough concentrations to produce LTP not only at the stimulated spine but at the adjacent spines into which they diffuse as a result of a lack of compartmentalization. The result is an overall increase in potentiation.[6]
In humans, many heritable disorders characterized by mental retardation are linked to mutations in genes important to the actin polymerization pathway. Williams syndrome, fragile X, fetal alcohol syndrome, and Patau syndrome have all been linked to these genes.[11] Neurons from people affected by these disorders show minimal dendritic arborization and underdeveloped spine structure, similar to neurons in animal models of molecular defects in actin polymerization.[1]
References
- Dillon, C., Goda, Y. (2005). The actin cytoskeleton: integrating form and function at the synapse. Annu. Rev. Neurosci., 28: 25-55.
- "Active Zone". Archived from the original on 2010-06-27. Retrieved 2010-04-27.
- Colicos MA, Collins BE, Sailor MJ, Goda Y. 2001. Remodeling of synaptic actin induced by photoconductive stimulation. Cell 107:605–16
- Fischer M, Kaech S, Knutti D, Matus A. 1998. Rapid actin-based plasticity in dendritic spines. Neuron 20:847–54
- Okamoto, K., Narayanan, R., Lee, S., Murata, K., Hayashi, Y., (2007) The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. PNAS, 104: 6418-6423.
- Meng, Y., Zhang, Y., Tregoubov, V., Janus, C., Cruz, I., et al. (2002). Abnormal spine morphology and enhanced LTP in LIMK1 knockout mice. Neuron, 35:121-133.
- Okamato, K. I., Nagai, T., Miyawaki, A., Hayashi, Y. (2004) Rapid and persistent modulation of actin dynamics regulates post-synaptic reorganization underlying bi-directional plasticity. Nature Neuroscience, 7:1104-1112.
- ZhangW, Benson DL. 2002. Developmentally regulated changes in cellular compartmentation and synaptic distribution of actin in hippocampal neurons. J. Neurosci. Res. 69:427–36
- Fukazawa, Y., Saitoh, Y., Ozawa, F., Ohta, Y., Mizuno, K., Inokochi, K. (2003). Hippocampal LTP is accompanied by enhanced f-actin content within dendritic spine that is essential for late LTP maintenance in vivo. Neuron, 38:447-460.
- Fischer, A., Sananbnesi, F., Schrick, C., Spiess, J., Radulovic, J. (2004). Distinct roles of hippocampul de novo protein synthesis and actin rearrangement in extinction of contextual fear. Journal of Neuroscience, 24:1962-1966.
- Chechlacz M, Gleeson JG. 2003. Is mental retardation a defect of synapse structure and function? Pediatr. Neurol. 29:11–17