Anticancer gene
Anticancer genes are genes that, when ectopically overexpressed, specifically destroy tumour cells without harming normal, untransformed cells. This cellular destruction can be due to a variety of mechanisms, such as apoptosis, mitotic catastrophe followed by apoptosis or necrosis, and autophagy. Anticancer genes emerged from studies on cancer cells in the late 1990s. Currently, there have been 291 anticancer genes discovered in the human genome. In order to be classified as an anticancer gene, the gene must have base substitutions leading to missense amino-acid changes, deletions, or insertions leading to frameshifts that alter the protein the gene codes for, increases and decreases in copy-number increases, or gene rearrangements leading to their deregulation.[1]
Anticancer genes as therapeutics
Cancer is classified as a group of diseases, all of which are characterized by uncontrolled cell proliferation.[2] In normal functioning cells, apoptosis is induced to avoid these proliferative events. However, these processes may continue on to become cancer in the event the processes become dysregulated. Epidemiological studies have shown cancer to be a leading cause of death worldwide [2] (Figure 1). Current advancements in therapeutics have led to a substantial increase in patient survival rates. Below is a non-comprehensive list of common anticancer genes.
Summary of anticancer genes
| Anti-cancer gene | Functional p53 required | Blocked by Bcl-2 | Caspases involved | Activated by phosphorylation | Engaging cell death pathway | Subcellular localization in cancer cells | Type of cell death |
|---|---|---|---|---|---|---|---|
| Apoptin | No | No | Yes | Yes | Intrinsic | Nucleus | Apoptosis |
| Brevinin-2R | Undetermined | Yes | No | Undetermined | Intrinsic | Cytoplasm | Autophagy |
| E4orf4 | No | No | No | Yes | Intrinsic | Nucleus, cytoplasm | Mitotic catastrophe |
| HAMLET | No | No | Yes | No | Intrinsic | Nucleus, ER, mitochondria | Apoptosis, autophagy |
| MDA-7 | No | Yes | Yes | No | Intrinsic | Receptor-binding, ER | Apoptosis |
| Noxa | No | Yes | Yes | Undetermined | Intrinsic | Mitochondria | Apoptosis |
| NS1 | No | No | No | Yes | Intrinsic | Cytoplasm | Apoptosis |
| ORCTL3 | Undetermined | Undetermined | Yes | Undetermined | Intrinsic | Plasma membrane, ER, golgi | Apoptosis |
| PAR-4 | No | No | Yes | Yes | Extrinsic, Intrinsic | Nucleus, ER, plasma membrane | Apoptosis |
| TRAIL | No | Yes | Yes | No | Extrinsic | Receptor-binding | Apoptosis |
Common anticancer gene examples
History
Apoptin was the first anticancer gene to be isolated. This gene comes from the single, circular minus-strand DNA found in the Chicken Anemia Virus (CAV) genome.[4] This virus belongs to the Gyrovirus genus, and is currently being studied as a new cancer therapeutic and diagnostic tool. This protein, also known as viral protein 3 (VP3) was isolated from chickens, and has been shown to cause PCD in transformed human cells.
Action
This protein encoded for by Apoptin has the specific capability of attacking transforming cells while leaving untransformed cells unharmed. Independent of p53, Apoptin induces apoptosis through an intrinsic, mitochondrial pathway. And unlike other PCD pathways, the pathway of Apoptin is independent of death receptors. In normal functioning cells, this 13.6-kDa protein resides in the cytoplasm, yet in cancerous cells, it travels to the nucleus via phosphorylation at the Thr-108 position via the mitogenic cyclin dependent kinase (CDK2). Additionally, this protein does not act alone. Several Apoptin-interacting molecules are needed in order for Apoptin to be fully functional. These molecules include, but not limited to, DNA, clyclinA-CDK2, and fas-associated death domain protein (FADD). Current apoptin therapeutic agents have been used to treat Lewis lung carcinomas, and osteosarcomas with future implications in treating liver cancers.[4]
History

Brevinin-2R is a peptide product isolated from the skin of the frog Rana ridibunda (Figure 2).[5] This non-hemolytic defensin has been shown to have preferential cytotoxicity towards various cancer cells including B-cell lymphoma, colon carcinomas, lung carcinomas, and breast adenocarcinoma.[6] Currently, this peptide and two of its analogs, Brevinin-2R-C and Brevinin-2R-D, are being explored for cancer drug development.[7] A phylogenetic analysis shows that Brevinin-2 is segregated into three major clades: A, B and C, where clade A contains the Brevining-2R homolog.[6]
Action
This 25 amino acid peptide, in contrast to the majority of peptides within the Brevinin family, has low hemolytic action.[7] Not only does the peptide have a reduced hemolytic action, it also is semi-selective towards cancer cells and leaves non cancerous cells largely unharmed. This peptide works as to prevent the progression of cancer by arresting the cell cycle at the G2/M phase, resulting in an induction of apoptosis.[7]
This defensin traditionally works as a part of the innate immune system, working as an antimicrobial defense.[8] However, this peptide is currently being studied as an anticancer peptide. Brevinin-2R works to trigger cell death by reducing the mitochondrial membrane potential resulting in lower cellular ATP levels while simultaneously increasing the concentration of reactive oxygen species.[8] Currently and somewhat unrelated, Brevinin-2R is being considered for diabetic treatments. In treating type II diabetes, or diabetes mellitus, Brevinins have been shown to promote insulin release. Finally, these peptides even have the capability to increase the rate of tissue regeneration, as seen with the frog in which Brevinin-2R was isolated from.[8]
History
Early region 4 open-reading-frame 4 (E4orf4) is an adenovirus protein of 14kDa which regulates growth in all stages of the adenovirus (Ad) infection. E4orf4 partners mainly with protein phosphatase 2A (PP2A) and Src kinases to induce cell death. Modeling of this protein reveals that it is likely made up of 3 α-helices with N- and C-terminal loops. It has a small stretch of amino acids in positions 66–75, which are highly basic, and likely are a place of nuclear and nucleolar targeting, as well as a place for Src kinases to bind.[9]
Action
E4orf4 is an important regulator of adenoviruses. Additionally, outside of the context of the virus, it causes programmed cell death both in the context of a healthy cellular environment, and cancer. E4orf4 is a key regulator of Ad by down-regulating both viral and cellular genes, which plays an important role in regulating the proliferation of the virus. In turn, the down-regulation also impacts the alternative splicing of the viral RNA and protein translation. In the absence of a viral infection, E4orf4 induces apoptosis in a p53 and caspase-independent manner; however, there is still communication between this pathway and the caspase-dependent apoptosis pathway. In the context of cancer, E4orf4 is even more efficient at inducing cell death than in healthy cells, which could be an important finding for potential cancer therapies. It has been discovered that the mechanisms behind the function of E4orf4 is closely associated with several other proteins including the B55 subunit of PP2A. E4orf4 binds to PP2A to reduce the phosphorylation of the DNA damage response (DDR) proteins. Consequently, this reduces the function of DDR and limits DNA repair. Many cancer cells have defects in the DDR pathways and targeting these cells with E4orf4 can potentially destroy the remaining DDR pathways, resulting in cancer cell death.[10]
The main mechanism behind the specificity of cancer cell targeting by E4orf4 is unknown but there are multiple hypotheses that scientists are considering: 1) The activation of the oncogenic state causes dormant apoptotic signals to be initiated and cause cell death to be more easily achieved by different signals. 2) There has been some indication that cancer cells become addicted to oncogenic pathways. E4orf4 may inhibit these pathways, causing cell death in cancer cells, but not normal cells. 3) E4orf4 may use oncogenes that have been activated in cancer cells, including Src, to cause cell death. 4) Cancer cells have disrupted cell cycle checkpoints and E4orf4 can take advantage of this by disrupting checkpoints in mitosis. 5) A Drosophila model demonstrated that E4orf4 can inhibit classical apoptosis in healthy tissues. It has been considered that this function of E4orf4 is lost in cancer cells causing a more effective killing of cells. 6) E4orf4 has been shown to cause structural changes in mitochondria, which could impact metabolic reprogramming and may affect cancer and healthy cells differently.[9]
History
HAMLET is known as an anticancer protein complex found in breast milk. One of the two molecules of this complex is multimeric alpha lactalbumin (MAL) (Figure 3), which was first discovered during a study in 1995 that investigated how breast milk affects bacteria transformed with lung cancer. This study found that transformed cells were selected for apoptosis at a much higher rate than the untransformed, healthy cells.[11] A later study in 2000, ascertained that oleic acid, a C18:1 fatty acid, is a cofactor that binds to MAL forming HAMLET. This complex, in a partially unfolded state, then displays apoptotic activity in cancer cells.[12]
Action
Apoptosis, or programmed cell death, can occur through activation of three different pathways, intrinsic, extrinsic, or tumor necrosis factor. HAMLET proceeds by both a multifaceted intrinsic pathway and the caspase cascade, a subsection of the TNF pathway, through targeting many different cell components.[13] First, after uptake by the cell, HAMLET proceeds to the mitochondria and depolarize the membranes at cytochrome c. Consequently, mitochondria dependent apoptosis factors are released as well as the caspase cascade is activated.[14] Second, proteasomes are targeted by HAMLET through a mechanism that is less understood. Research does suggest that HAMLET directly binds to the proteasome leading to its inhibition.[15] Third, HAMLET has been found to target the nucleus, specifically histones. HAMLET irreversibly binds to histones leading to the inactivation of transcription and chromatin condensation, which inevitably causes apoptosis.[16] Lastly, studies show that cells treated by HAMLET exhibit behaviors common to macroautophagy. This includes presence of cytoplasmic vacuoles, double-membrane vesicles, and a dose-dependent decrease in ATP levels.[13]
History
Melanoma differentiation associated gene-7 (mda-7), and also known as IL-24, was discovered in the mid-1900s using subtraction hybridization. mda-7 is classified in the interleukin IL-10 family because of similar structure and amino acid sequence to other interleukins in that class, the chromosomal location (human chromosome 1q32-33),[17] and the shared properties it has with cytokines. Protein structural studies reveal that it is a dimer and glycolsylated. It has been found that its expression is either not present or present at very low levels in tumor cells, including advanced stage melanoma and metastatic disease, compared to normal non-transformed cells. Multiple studies within the past 15 years have demonstrated that increasing mda-7 expression in tumor cells results in growth arrest and cell death in many different cell lines. When mda-7 is over-expressed in normal cells, no change in growth or cell viability is detected. mda-7 is also considered a radio-sensitizing cytokine because it generates a reactive oxygen species and causes stress in endoplasmic reticulum.[18] mda-7 has been used in several clinical trials because of its ability to induce apoptosis, prevent tumor angiogenesis, cause immune-regulation, and increase radiation lethality. It was seen in one Phase I clinical trial that injecting mda-7 via an adenovirus directly into a tumor resulted in safe tumor regulation and immune activation.[18]
Action
mda-7 interacts with two of the type II cytokine hetero-dymeric receptor complexes IL-20R1/IL-20R2 and IL-22R1/IL-20R2. It has been seen that in some contexts, mda-7 activates STAT transcription factors. However, the STAT pathway is not always activated and is not required for mda-7 cell growth arrest and cell death. mda-7 can be placed into tumor cell lines via transfection or adenovirus-transduction; it has been seen that following this, apoptosis is induced only in the tumor cells and results in no toxicity in the healthy cells.[17] Its function as a tumor suppressor is not fully understood, but it has been observed that in the context of melanoma, mda-7 expression is drastically decreased. While there are no official studies published backing this claim, it is thought that mda-7 could potentially act as a paracrine factor, be involved in signaling short-range, and immune function in skin. mda-7 is also thought to have a pro-inflammatory purpose. It is also possible that mda-7 induces cytokine secretion, which causes antigen-presenting cells to present tumor antigens, resulting in an immune response against tumors. It has also been discovered that mda-7, and its translated protein MDA-7, interacts with kinases including serine/threonine protein kinase (PKR).[17] Further studies will need to be performed to better understand the mechanisms of mda-7 action.
History
Noxa, isolated from mice, is a member of the Bcl-2 family and is able to regulate cell death through a variety of intracellular stress signals.[19] Having been discovered nearly three decades ago in 1990 by Hijikata et al., this gene product was isolated this protein from an adult T-cell leukemia (ATL) library[20] This gene, and its protein in which it encodes for, has been studied as a potential therapeutic in chronic lymphocytic leukemia (CLL), the most common leukemia found in adults in the Western world.[19] In humans, the Noxa homologue is known as APR/PMAIP1.[20]
Action
Upon receiving intrinsic death signals, the gene NOXA encodes for the protein Noxa through a three-exon transcript.[20] This protein binds to anti-apoptotic proteins resulting in these proteins' inhibition.[19] As a p53 inducible gene, NOXA is transcribed and translated to Noxa in response to DNA damage and hypoxia induced apoptosis.[19] A constitutive gene found in the brain, thymus, spleen, and several other organs, it initiates apoptosis through Bax-mediated mitochondrial-dysfunction through the inhibition of the Bcl2 family's antiapoptotic members.[20] Through gene knockout studies, it was shown that double deficient Noxa there was no spontaneous tumor development as commonly observed with knockout of p53.[20] Noxa has been shown to be involved in the maintenance of memory CD4+ T Th1/Th2 cell homeostasis where in the absence of Noxa, Th2 memory T-cell death results.[20]
History
In the 1960s rodent parvovirus was discovered by Dr. Helene Toolan to have an oncosuppressive activity.[21][22][23][24][25] However, the specific gene found in the parvovirus genome, which is called NS1, that causes the oncosuppressive activity was not characterized until later. NS1 is a small protein (only 672 amino acids) with 5 distinct domains that exert different functions that inevitably lead to apoptosis and cell death. NS1 activates cell death through two different pathways, apoptosis/lysosomal-like programmed cell death and necrosis/cytolysis.[26]
Action
NS1 is considered a regulatory protein due to its activity in transcription, translation, and protein-protein interactions, which allows the parvovirus to replicate unhindered. However, scientists are primarily interested in utilizing its cytolytic activity since this has been proven to be active in cancerous cells. The first way NS1 propagates cell death through cytolysis is by interrupting the cell cycle at the S/G2 junction, causing a stress response in the cell. Specifically, NS1 interacts with many molecules and compounds important in the transition and inhibits their activity. When NS1 expression reaches a certain threshold, the triggered stress response finally causes caspase 3/9-mediated programmed cell death.[26] Another way that NS1 causes cytolysis is through degradation of the cytoskeleton of the cell. NS1 specifically targets and degrades the microfilament tropomyosin using casein kinase II, actin filaments through activation of actin-severing protein gelsolin, and vimentin through an unknown mechanism.[27][28][29] The last NS1-mediated mechanism of cytolysis involves the depolarization of the mitochondria. This results in the release of many reactive oxygen species, causing DNA damage. When DNA is damaged, a DNA damage response occurs, which in this case results in cell death.[30]
History
Organic Cation Transporter Like-3 (ORCTL3) was first discovered as a result of a large-scale DNA sequencing project in search of genes with a tumor-specific apoptosis activity.[31] The name ORCTL3 was decided upon because of its structural homology to proteins belonging to the family of organic cation transporters.[32] However, the name is a misnomer as after examining the properties of ORCTL3, it was revealed that ORCTL3 is a transporter for urate. The ORCTL3 gene spans around 12 kb of genomic DNA and consists of ten exons. It was shown that the 2.4 kb transcript of this gene is universally expressed in all human tissues. Additionally, ORCTL3 transfection into numerous tumorigenic cells induced apoptosis, while normal and primary cells remained healthy.[33]
Action
ORCTL3 is a 90 kDa protein composed of 351 amino acids.[34][35] It is suggested that the protein spans the cell membrane several times, based on computational methods.[36] Overexpressed ORCTL3 is localized to the endoplasmic reticulum (ER), Golgi and the plasma membrane but not to mitochondria.[33] ORCTL3 was identified as the first high-affinity nicotinate exchanger in kidneys and intestine. Nicotinate is an essential vitamin (Vitamin B3) that is involved in NAD+ synthesis, which in turn is important for energetic processes, signal transduction pathways, and the activation of the NAD+ -dependent histone deacetylase SIRT1. ORCTL3 has been shown to be activated for apoptosis induction in renal cells in vitro, in vivo and ex vivo. For its apoptosis effect ORCTL3 targets stearoyl-CoA desaturase (SCD), an enzyme that introduces a double bond in the fatty acid stearic acid.[37] The fact that SCD is commonly overexpressed in cancer and oncogene transformed cells might explain the tumor-specificity of ORCTL3 to some extent, however, the existence of other additional targets of ORCTL3 cannot formally be ruled out.
History
Prostate apoptosis response-4 (Par-4) is a tumor suppressor protein with a pro-apoptotic function. Par-4 was first discovered in rat prostate cancer cells as part of an effort determined in discovering genes that were induced in response to increased Ca2+ in cells, although it is now known to be ubiquitously expressed in a wide variety of tissues across many different species.[38] The Par-4 gene is located on the minus strand of chromosome 12q21.2, spanning 99.06 kb of DNA and containing seven exons and six introns. Par-4 is known to be downregulated in certain terminally differentiated cells such as neurons, specific retinal cells, and smooth muscle cells as well as in certain cancer cells such as renal cancers, neuroblastoma, and leukemia.[39][40] Par-4 has also been shown to be generally higher in dying cells, consistent with its pro-apoptotic functions.
Action
Par-4 is a 38 kDa multi-domain protein composed of about 340 amino acids. Conserved domains among human, mouse, and rat homologs include the leucine zipper (LZ) domain at the C-terminal region, two nuclear localization sequences, NLS1 and NLS2, in the N-terminal region, and a nuclear export sequence within the LZ domain.[41] Although Par-4 mutations are rare, it was identified that an A to T point mutation affecting residue 189 localized in exon 3 causes premature termination of Par-4 in human endometrial carcinoma.[42] Knockout of Par-4 in mice leads to the development of spontaneous tumors in various tissues revealed by increased proliferative response of peripheral T cells, inhibition of apoptosis, increased NF-κB activity, and decreased JNK activity.[43] Par-4 overexpression is sufficient to induce apoptosis in most cancer cells in the absence of a second apoptotic signal, but does not induce apoptosis in normal or immortalized cells.[41][44][45]
The anticancer function of Par-4 is achieved by two distinct means: activating the molecular components of the cell-death machinery and inhibiting pro-survival factors. One essential apoptotic function of Par-4 is inhibiting the NF-κB pathway, which is a key contributing factor in many tumors and prevents cell death by activating the expression of pro-survival genes. Par-4 also assists in PCD by enabling the trafficking of specific ligands such and cell surface death receptors, such as FasL and Fas, respectively, to the plasma membrane thus activating the extrinsic death pathway. Overexpression of Par-4 selectively induces apoptosis in cancer cells, attributed to the selective activation via phosphorylation of the T155 residue by protein kinase A (PKA).[46] It has been shown that two events are required for Par-4 activation: nuclear entry and phosphorylation by PKA.
History
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (Figure 5) is a member of the tumor necrosis factor (TNF) family that also includes Fas ligands, TNFα, and TL1A. It was discovered in 1995 by Wiley et al. and then further characterized in 1996 by Pitti et al. The former study discovered that TRAIL is localized to surfaces of cells in most human tissues, excluding the brain, liver, and testes,[47] while the latter study was able to elicit that the protein is a type II membrane protein that can also be cleaved into a soluble form.[48]
Action
The intrigue surrounding TRAIL is all due to this protein's ability both in vivo and in vitro to specifically target tumor cells for apoptosis while leaving healthy cells intact. This activity proceeds by both the intrinsic and extrinsic pathway. First, the homotrimer of TRAIL binds three molecules of either TRAIL-receptor 1 or 2, which are transmembrane proteins that contain a cytoplasmic death domain. Once TRAIL is bound, Fas, caspase-8, and caspase-10 associate with the death domain forming death-inducing signaling complex (DISC) that proceeds through two different mechanisms depending on the cell type. In one cell type, DISC can directly activate the effector caspase leading to apoptosis, while in the other the complex activates a bcl-2-mediated pathway in a similar fashion as HAMLET that results in the release of cytochrome c from the mitochondria, which then causes the activation of effector caspase. The latter mechanism is the focus of many oncogenic therapies because p53, the tumor suppressor gene, activates the same pathway. Since cancer is commonly caused by the inactivation of p53, TRAIL could mediate this effect by still activating the apoptotic pathway.[49]
History
TP-53 (Figure 6) is a gene that encodes for the protein p53; this protein is a tumor suppressor. p53 was discovered in 1979 stemming from a study involving cancer immunology and the role of viruses in some cancers. The protein was so named because it was measured to have a weight of 53 kDa. This study was conducted by David Philip Lane and technician Alan K. Roberts, in Lionel V. Crawford's lab in London. It was seen in this study that p53 could bind to viral tumor antigens. This information was corroborated during the same year when a separate study found that p53 had immunoreactivity with serum from tumors containing antibodies. This later study was run by Daniel I. H. Linzer and Arnold J. Levine out of Princeton University. Further papers came out around the same time all mentioning the discovery of a tumor suppressing protein. While p53 was first officially identified in 1979, many labs in previous years had come across the same protein, without knowing what it was. In the mid-1970s, a scientist by the name of Peter Tegtmeyer happened upon a protein with an approximate size of 50 kDa. However, because he was focusing his studies on SV40, a tumor-causing virus affecting monkeys and humans, he did not pay much attention to this protein.[50]
Action
The p53 protein is a tumor-suppressing transcription factor (TF), which can recognize when there is an alteration in a cell's DNA caused by factors including chemical toxins, radiation, ultraviolet (UV) rays, and other damaging agents.[51] Crucially, p53 plays a role in determining whether the damaged genetic material in the cell can be repaired, or if the cell should be destroyed through apoptosis.[52][53] The individual topologically associating domains (TADs) target different genes and unique effector pathways. It has been observed that inactivating both of the TADs detrimentally affects the ability of p53 to suppress tumor growth and interact with target genes. When only one TAD is inactivated, p53 can still suppress specific tumors; however, it can no longer successfully engage in transactivation. The C-terminal domain (CTD) is an intrinsically disordered domain (IDD), which can take on different conformations depending on what it is binding with and is a location of many post-translational modifications, resulting in its ability to regulate p53 function depending on what it is bound to and what modifications are linked with the CTD. This domain also aids in the binding of the central DNA-binding domain (DBD) to specific DNA sequences; the CTD is a positive regulator of DNA binding and stabilizes the interaction of the DNA with the DBD.[51] p53 is unique as a transcription factor in that it can recognize and bind response elements (RE) in many different environments and doesn't need other transcription factors to cooperatively bind with it like many other TFs.[51]
Mutations in the p53 pathway have been observed in almost all cancer types including breast cancer, bladder cancer, lung cancer, ovarian cancer, cholangiocarcinoma, head and neck squamous cell carcinoma, melanoma, wilms tumor, and other cancers often due to a single point mutation in p53.[52][53] Li-Fraumeni Syndrome is a condition linked to inherited mutations, at least 140 mutations, in the TP-53 gene. This condition largely increases the risk of developing cancers like breast cancer, bone cancer, and soft tissue sarcomas. Specifically, this impacts children and young adults. A majority of these mutations in the TP-53 gene are single amino acid changes, but other mutations cause a small portion of the DNA to be absent. This leads to a faulty p53 protein that fails to recognize DNA damage in cells, control cell growth, and initiate apoptosis in cells with damaged DNA. Consequently, cells containing erroneous DNA can uncontrollably divide.[52]
Common misconceptions
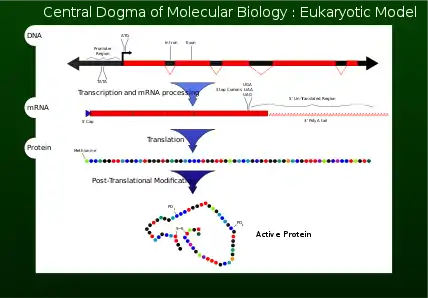
Often, genes are confused with the proteins in which they code for (Figure 7). Genes are composed of nucleotides, while proteins are composed of amino acids. The genes serve as codes and blueprints to create either proteins of interest, or various non-coding ribonucleic acids (ncRNAs), which exhibit various effects, such as working to prevent cancer within cells.
See also
- Oncogene
- Gene therapy for cancer
- Tumour suppressor gene
- Cell Cycle
- Cell Cycle Checkpoints
- Genes
- Proteins
- Apoptosis
- HAMLET
- Par-4
References
- Futreal, P. Andrew (2009). "A census of human cancer genes". Nature Reviews Cancer. 4 (3): 177–183. doi:10.1038/nrc1299. PMC 2665285. PMID 14993899.
- Olugbami, Jeremiah. "A comparative assessment of antiproliferative properties of resveratrol and ethanol leaf extract of Anogeissus leiocarpus (DC) Guill and Perr against HepG2 hepatocarcinoma cells". BMC Complementary and Alternative Medicine. 17: 1–11.
- Grimm, Stefan; Noteborn, Mathieu (2010-02-01). "Anticancer genes: inducers of tumour-specific cell death signalling". Trends in Molecular Medicine. 16 (2): 88–96. doi:10.1016/j.molmed.2009.12.002. ISSN 1471-4914. PMID 20138582.
- Los, M (2009). "Apoptin, a tumor-selective killer". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1793 (8): 1335–1342. doi:10.1016/j.bbamcr.2009.04.002. PMID 19374922.
- Zuse, Anne. "Brevinin-2R, a peptide inducing semi-selectively cancer cell death by a mechanism involving the mitochondrial death pathway". Cellular and Molecular Biology.
- Ghavami, Saeid (2008). "Brevinin-2R semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway". Cellular and Molecular Medicine. 12 (3): 1005–1022. doi:10.1111/j.1582-4934.2008.00129.x. PMC 4401144. PMID 18494941.
- Jamadi, Robab (2020). "Anticancer Activity of Brevinin-2R Peptide and its Two Analogues Against Myelogenous Leukemia Cell Line as Natural Treatments: An In Vitro Study". International Journal of Peptide Research and Therapeutics. 26 (2): 1013–1020. doi:10.1007/s10989-019-09903-6. S2CID 199407384.
- Zohrab, Fatemeh. "Biological Properties, Current Applications and Potential Therapeutic Applications of Brevinin Peptide Superfamily". Nature Public Health Emergency Collection. 25: 39–48.
- Kleinberger, Tamar (2015-05-07). "Mechanisms of Cancer Cell Killing by the Adenovirus E4orf4 Protein". Viruses. 7 (5): 2334–2357. doi:10.3390/v7052334. ISSN 1999-4915. PMC 4452909. PMID 25961489.
- Brestovitsky, Anna; Nebenzahl-Sharon, Keren; Kechker, Peter; Sharf, Rakefet; Kleinberger, Tamar (2016-02-11). "The Adenovirus E4orf4 Protein Provides a Novel Mechanism for Inhibition of the DNA Damage Response". PLOS Pathogens. 12 (2): e1005420. doi:10.1371/journal.ppat.1005420. ISSN 1553-7374. PMC 4750969. PMID 26867009. S2CID 14919067.
- Håkansson, A; Zhivotovsky, B; Orrenius, S; Sabharwal, H; Svanborg, C (1995-08-15). "Apoptosis induced by a human milk protein". Proceedings of the National Academy of Sciences of the United States of America. 92 (17): 8064–8068. Bibcode:1995PNAS...92.8064H. doi:10.1073/pnas.92.17.8064. ISSN 0027-8424. PMC 41287. PMID 7644538.
- Svensson, M.; Håkansson, A.; Mossberg, A.-K.; Linse, S.; Svanborg, C. (2000-04-11). "Conversion of α-lactalbumin to a protein inducing apoptosis". Proceedings of the National Academy of Sciences of the United States of America. 97 (8): 4221–4226. Bibcode:2000PNAS...97.4221S. doi:10.1073/pnas.97.8.4221. ISSN 0027-8424. PMC 18203. PMID 10760289.
- Hallgren, Oskar; Aits, Sonja; Brest, Patrick; Gustafsson, Lotta; Mossberg, Ann-Kristin; Wullt, Björn; Svanborg, Catharina (2008), Bösze, Zsuzsanna (ed.), "Apoptosis and Tumor Cell Death in Response to HAMLET (Human α-Lactalbumin Made Lethal to Tumor Cells)", Bioactive Components of Milk, Advances in Experimental Medicine and Biology, New York, NY: Springer, vol. 606, pp. 217–240, doi:10.1007/978-0-387-74087-4_8, ISBN 978-0-387-74087-4, PMID 18183931, retrieved 2020-10-18
- Köhler, Camilla (2001-11-23). Mechanisms of apoptosis induced by a protein complex isolated from human milk : With focus on the role of mitochondria. Institutet för miljömedicin (IMM) / Institute of Environmental Medicine. ISBN 978-91-7349-048-1.
- Gustafsson, Lotta (2005). HAMLET - In vivo effects and mechanisms of tumor-cell death (thesis/doccomp thesis). Lund University.
- Düringer, Caroline; Hamiche, Ali; Gustafsson, Lotta; Kimura, Hiroshi; Svanborg, Catharina (2003-10-24). "HAMLET Interacts with Histones and Chromatin in Tumor Cell Nuclei". Journal of Biological Chemistry. 278 (43): 42131–42135. doi:10.1074/jbc.M306462200. ISSN 0021-9258. PMID 12888554. S2CID 34301355.
- GUPTA, P; SU, Z; LEBEDEVA, I; SARKAR, D; SAUANE, M; EMDAD, L; BACHELOR, M; GRANT, S; CURIEL, D; DENT, P (September 2006). "mda-7/IL-24: Multifunctional cancer-specific apoptosis-inducing cytokine". Pharmacology & Therapeutics. 111 (3): 596–628. doi:10.1016/j.pharmthera.2005.11.005. ISSN 0163-7258. PMC 1781515. PMID 16464504.
- Dent, Paul; Yacoub, Adly; Hamed, Hossein A.; Park, Margaret A.; Dash, Rupesh; Bhutia, Sujit K.; Sarkar, Devanand; Gupta, Pankaj; Emdad, Luni; Lebedeva, Irina V.; Sauane, Moira (September 2010). "MDA-7/IL-24 as a cancer therapeutic: from bench to bedside". Anti-Cancer Drugs. 21 (8): 725–731. doi:10.1097/CAD.0b013e32833cfbe1. ISSN 0959-4973. PMC 2915543. PMID 20613485.
- Zhang, L-N (2013). "A review of the role of Puma, Noxa, and Bim in the tumorigenesis therapy and drug resistance of chronic lymphocytic leukemia". Nature. 20 (1): 1–7. doi:10.1038/cgt.2012.84. PMID 23175245. S2CID 7183342.
- Ploner, C. "Noxa: at the tip of the balance between life and death". Nature. 27: S84–S92.
- Toolan, H.W., Saunders, E.L., Southam, C.M., Moore, A.E. and Levin, A.G. (1965) H-l virus viremia in the human. Proc. Sot. Exp. Biol. Med. 119, 711-715.
- Toolan, H.W., Rhode, S.L. and Gierthy, J.F. (1982) Inhibition of 7, 12-dimethylbenz(a)anthracene-induced tumors in Syrian hamsters by prior infection with H-l parvovirus. Cancer Res. 42,2552- 25.55.
- Toolan, H.W. and Ledinko, N. (1968) Inhibition by H-l virus of the incidence of tumors produced by adenovirus 12 in hamsters. Virology 35, 475478.
- Toolan, H.W. (1967) Lack of oncogenic effect of the H-viruses. Nature 214, 1036.
- Toolan, H.W. and Ledinko, N. (1965) Growth and cytopathogenicity of H-viruses in human and simian cell cultures. Nature 208, 8 12-8 13.
- Nüesch, Jürg P. F.; Rommelaere, Jean (2014). "Tumor Suppressing Properties of Rodent Parvovirus NS1 Proteins and Their Derivatives". Anticancer Genes. Advances in Experimental Medicine and Biology. Vol. 818. London: Springer. pp. 99–124. doi:10.1007/978-1-4471-6458-6_5. ISBN 978-1-4471-6457-9. PMID 25001533.
- Christensen J, Cotmore SF, Tattersall P (1995) Minute virus of mice transcriptional activator protein NS1 binds directly to the transactivation region of the viral P38 promoter in a strictly ATP-dependent manner. J Virol 69:5422–5430
- Nuesch JP, Bar S, Rommelaere J (2008) Viral proteins killing tumor cells: new weapons in the fight against cancer. Cancer Biol Ther 7:1374–1376
- Nuesch JP, Lachmann S, Rommelaere J (2005) Selective alterations of the host cell archi- tecture upon infection with parvovirus minute virus of mice. Virology 331:159–174
- Hristov G, Kramer M, Li J, El-Andaloussi N, Mora R, Daeffler L et al (2010) Through its nonstructural protein NS1, parvovirus H-1 induces apoptosis via accumulation of reactive oxygen species. J Virol 84:5909–5922
- Murata, Yasushi; Tamari, Mayuml; Takahashl, Takashi; Horio, Yoshltsugu; Hlbi, Kenji; Yokoyama, Shiro; Inazawa, Johjl; Yamakawa, Kazuhiro; Ogawa, Akimi; Takahashi, Toshitada; Nakamura, Yusuke (1994-08-01). "Characterization of an 800 kb region at 3p22-p21.3 that was homozygously deleted in a lung cancer cell line". Human Molecular Genetics. 3 (8): 1341–1344. doi:10.1093/hmg/3.8.1341. ISSN 0964-6906. PMID 7987312.
- Nishiwaki, T.; Daigo, Y.; Tamari, M.; Fujii, Y.; Nakamura, Y. (1998). "Molecular cloning, mapping, and characterization of two novel human genes, ORCTL3 and ORCTL4, bearing homology to organic-cation transporters". Cytogenetic and Genome Research. 83 (3–4): 251–255. doi:10.1159/000015197. ISSN 1424-8581. PMID 10072596. S2CID 9118091.
- Irshad, S.; Mahul-Mellier, A.-L.; Kassouf, N.; Lemarie, A.; Grimm, S. (June 2009). "Isolation of ORCTL3 in a novel genetic screen for tumor-specific apoptosis inducers". Cell Death & Differentiation. 16 (6): 890–898. doi:10.1038/cdd.2009.21. ISSN 1476-5403. PMC 2683172. PMID 19282870.
- Lee, Woon Kyu; Hwang, Ji-Sun; Yun, Cheol-Heui; Cha, Seok Ho (December 2007). "Identification of a kidney-specific mouse organic cation transporter like-1 (mOCTL1)". Experimental & Molecular Medicine. 39 (6): 787–795. doi:10.1038/emm.2007.85. ISSN 2092-6413. PMID 18160849. S2CID 23950699.
- Bahn, Andrew; Hagos, Yohannes; Reuter, Stefan; Balen, Daniela; Brzica, Hrvoje; Krick, Wolfgang; Burckhardt, Birgitta C.; Sabolić, Ivan; Burckhardt, Gerhard (2008-06-13). "Identification of a New Urate and High Affinity Nicotinate Transporter, hOAT10 (SLC22A13)". Journal of Biological Chemistry. 283 (24): 16332–16341. doi:10.1074/jbc.M800737200. ISSN 0021-9258. PMID 18411268. S2CID 5522658.
- Kyte, Jack; Doolittle, Russell F. (1982-05-05). "A simple method for displaying the hydropathic character of a protein". Journal of Molecular Biology. 157 (1): 105–132. doi:10.1016/0022-2836(82)90515-0. ISSN 0022-2836. PMID 7108955.
- AbuAli, G.; Chaisaklert, W.; Stelloo, E.; Pazarentzos, E.; Hwang, M.-S.; Qize, D.; Harding, S. V.; Al-Rubaish, A.; Alzahrani, A. J.; Al-Ali, A.; Sanders, T. a. B. (March 2015). "The anticancer gene ORCTL3 targets stearoyl-CoA desaturase-1 for tumour-specific apoptosis". Oncogene. 34 (13): 1718–1728. doi:10.1038/onc.2014.93. ISSN 1476-5594. PMC 4119473. PMID 24769897.
- El-Guendy, Nadia; Rangnekar, Vivek M (2003-02-01). "Apoptosis by Par-4 in cancer and neurodegenerative diseases". Experimental Cell Research. 283 (1): 51–66. doi:10.1016/S0014-4827(02)00016-2. ISSN 0014-4827. PMID 12565819.
- Cook, Jason; Krishnan, Sumathi; Ananth, Subbian; Sells, Stephen F.; Shi, Yang; Walther, McClellan M.; Linehan, W. Marston; Sukhatme, Vikas P.; Weinstein, Michael H.; Rangnekar, Vivek M. (February 1999). "Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma". Oncogene. 18 (5): 1205–1208. doi:10.1038/sj.onc.1202416. ISSN 1476-5594. PMID 10022126. S2CID 10990391.
- Kögel, D.; Reimertz, C.; Mech, P.; Poppe, M.; Frühwald, M. C.; Engemann, H.; Scheidtmann, K. H.; Prehn, J. H. M. (December 2001). "Dlk/ZIP kinase-induced apoptosis in human medulloblastoma cells: requirement of the mitochondrial apoptosis pathway". British Journal of Cancer. 85 (11): 1801–1808. doi:10.1054/bjoc.2001.2158. ISSN 1532-1827. PMC 2363987. PMID 11742505.
- El-Guendy, Nadia; Zhao, Yanming; Gurumurthy, Sushma; Burikhanov, Ravshan; Rangnekar, Vivek M. (2003-08-15). "Identification of a Unique Core Domain of Par-4 Sufficient for Selective Apoptosis Induction in Cancer Cells". Molecular and Cellular Biology. 23 (16): 5516–5525. doi:10.1128/MCB.23.16.5516-5525.2003. ISSN 0270-7306. PMC 166354. PMID 12897127.
- Moreno-Bueno, Gema; Fernandez-Marcos, Pablo J.; Collado, Manuel; Tendero, Mercedes J.; Rodriguez-Pinilla, Socorro M.; Garcia-Cao, Isabel; Hardisson, David; Diaz-Meco, Maria T.; Moscat, Jorge; Serrano, Manuel; Palacios, Jose (2007-03-01). "Inactivation of the Candidate Tumor Suppressor Par-4 in Endometrial Cancer". Cancer Research. 67 (5): 1927–1934. doi:10.1158/0008-5472.CAN-06-2687. ISSN 0008-5472. PMID 17332319.
- García-Cao, Isabel; Duran, Angeles; Collado, Manuel; Carrascosa, Maria J; Martín-Caballero, Juan; Flores, Juana M; Diaz-Meco, Maria T; Moscat, Jorge; Serrano, Manuel (2005-06-01). "Tumour-suppression activity of the proapoptotic regulator Par4". EMBO Reports. 6 (6): 577–583. doi:10.1038/sj.embor.7400421. ISSN 1469-221X. PMC 1369092. PMID 15877079.
- Chakraborty, Mala; Qiu, Shirley Guofang; Vasudevan, Krishna Murthi; Rangnekar, Vivek M. (2001-10-01). "Par-4 Drives Trafficking and Activation of Fas and FasL to Induce Prostate Cancer Cell Apoptosis and Tumor Regression". Cancer Research. 61 (19): 7255–7263. ISSN 0008-5472. PMID 11585763.
- Nalca, Aysegul; Qiu, Shirley Guofang; El-Guendy, Nadia; Krishnan, Sumathi; Rangnekar, Vivek M. (1999-10-15). "Oncogenic Ras Sensitizes Cells to Apoptosis by Par-4". Journal of Biological Chemistry. 274 (42): 29976–29983. doi:10.1074/jbc.274.42.29976. ISSN 0021-9258. PMID 10514481. S2CID 2551093.
- Gurumurthy, Sushma; Goswami, Anindya; Vasudevan, Krishna Murthi; Rangnekar, Vivek M. (2005-02-01). "Phosphorylation of Par-4 by Protein Kinase A Is Critical for Apoptosis". Molecular and Cellular Biology. 25 (3): 1146–1161. doi:10.1128/MCB.25.3.1146-1161.2005. ISSN 0270-7306. PMC 544017. PMID 15657440.
- Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995;3:673–82
- Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 1996;271:12687–90.
- Carlo-Stella, Carmelo; Lavazza, Cristiana; Locatelli, Alberta; Viganò, Lucia; Gianni, Alessandro M.; Gianni, Luca (2007-04-15). "Targeting TRAIL Agonistic Receptors for Cancer Therapy". Clinical Cancer Research. 13 (8): 2313–2317. doi:10.1158/1078-0432.CCR-06-2774. ISSN 1078-0432. PMID 17438088. S2CID 7424982.
- "The Discovery of p53 Protein | The Embryo Project Encyclopedia". embryo.asu.edu. Retrieved 2020-10-05.
- Sullivan, Kelly D.; Galbraith, Matthew D.; Andrysik, Zdenek; Espinosa, Joaquin M. (January 2018). "Mechanisms of transcriptional regulation by p53". Cell Death & Differentiation. 25 (1): 133–143. doi:10.1038/cdd.2017.174. ISSN 1476-5403. PMC 5729533. PMID 29125602.
- "TP53 gene: MedlinePlus Genetics". medlineplus.gov. Retrieved 2020-10-04.
- Li, Lijuan; Wu, Jian; Sima, Xiutian; Bai, Peng; Deng, Wei; Deng, Xueke; Zhang, Lin; Gao, Linbo (2013-03-17). "Interactions of miR-34b/c and TP-53 polymorphisms on the risk of nasopharyngeal carcinoma". Tumor Biology. 34 (3): 1919–1923. doi:10.1007/s13277-013-0736-9. ISSN 1010-4283. PMID 23504554. S2CID 17155357.