Neutral lipid storage disease
Neutral lipid storage disease (also known as Chanarin–Dorfman syndrome) is a congenital autosomal recessive disorder characterized by accumulation of triglycerides in the cytoplasm of leukocytes[1], (Jordan’s Anomaly) muscle, liver, fibroblasts, and other tissues. It commonly occurs as one of two subtypes, cardiomyopathic neutral lipid storage disease (NLSD-M), or ichthyotic neutral lipid storage disease (NLSD-I) which is also known as Chanarin–Dorfman syndrome), which are characterized primarily by myopathy and ichthyosis, respectively. Normally, the ichthyosis that is present is typically non-bullous congenital ichthyosiform erythroderma which appears as white scaling.
| Neutral lipid storage disease | |
|---|---|
| Other names | Chanarin–Dorfman syndrome |
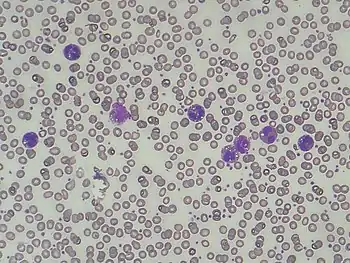 | |
| Presence of lipid vacuoles in granulocytes in Chanarin-Dorfman syndrome (also known as Jordans' anomaly) | |
It has been associated genetically with mutations in the CGI58 gene, (for NLSD-I), or the ATGL gene (for NLSD-M.)[1][2][3]
Cause
Neutral lipid storage disease is caused by the abnormal and excessive accumulation of lipids in certain bodily tissues, including the liver, the heart, and muscle.[4] Normally, these lipids are stored as lipid droplets and are normally used for metabolism, cell signaling and trafficking of vesicles.[5] Neutral lipid storage disease is a disease that is diagnosed with the simultaneous occurrence of myopathy and/or ichthyosis. Myopathy is defined as a disease of the muscle tissue. Ichthyosis is a skin related disease in which the skin becomes very scaly, thick, and dry.
Genetics
Neutral lipid storage disease (NLSD) occurs in one of two genetic and clinical subtypes. Both subtypes are autosomal recessive disorders, meaning that a mutant allele must be inherited from both parents in order to cause disease.
Subtype I: Neutral Lipid Storage Disease with Myopathy (NLSD-M), is caused by a mutation in the PNPLA2 gene, which reduces the normal expression orfunction of the ATGL protein. PNPLA2 is located on chromosome 11.[6] ATGL is an enzyme involved in catabolism of triglycerides (long-term fat storage) into fatty acids (short-term fat storage) within the body In the absence of fully functional ATGL, triglycerides accumulate in the bloodstream and bodily tissues. Interestingly, individuals with NLSD are not typically obese. It has been proposed that the assimilation, rather than degradation of triglycerides is the main factor in fat accumulation in adipose cells.[7] In the absence of functional ATGL, triglycerides accumulate in the bloodstream and bodily tissues. Interestingly, individuals with NLSD are not typically obese. It has been proposed that the assimilation, rather than degradation of triglycerides is the main factor in fat accumulation in adipose cells.[8] Patients with NLSD-M display progressive skeletal myopathy and severe cardiomyopathy in ~40% of cases.[9] The pathophysiology and mechanistic basis of myopathy arising from deficits is lipid metabolism is not yet known.
Subtype II: Neutral Lipid Storage Disease with Ichthyosis (NLSD-I), or Chanarin-Dorfman syndrome, is caused by a mutation in the CGI-58 protein. CGI-58 is a coactivator of ATGL, and together these proteins sustain blood lipid levels between meals.[10][11] Disruption of CGI-58 gives rise to the symptoms of ichthyosis, a dermatological condition in which skin becomes very scaly, thick and dry.
Ichthyosis is an inherited disorder, like NLSD, and has been found to be genetically linked in some situations with the PNPLA2 gene. Therefore, sometimes when there is a mutation in the PNPLA2 gene, the linked allele of CG-58 is mutated as well. This specific gene, however, does not produce ichthyosis on its own. Ichthyosis can be diagnosed individually in a patient and the genetic cause for unlinked ichthyosis is different from the gene linked with PNPLA2. Keep in mind these disorders may occur individually within a patient as well.
Pathophysiology
The functional changes that occur with this disease are mostly metabolic. Accumulation of triglycerides in the body without an efficient mode for catabolism is thought to lead to the eventual symptoms of this disease. Upon digestion and absorption of fat by the small intestine, triglycerides are combined with vitamins and cholesterol to form chylomicrons. Chylomicrons travel from the intestine into the lymph system before entering the bloodstream. Enzymatic catalysis of chylomicrons by lipases in the bloodstream enables the uptake of lipids and fatty acids by cells. In individuals with NLSD, their triglycerides are not catabolized in the blood, and cells accumulate partially processed lipid droplets over time, which may lead to dysfunction in absorbing tissues. In affected individuals, muscle cells, fibroblasts, and leukocytes appear to be prone to the excessive accumulation of triglycerides as lipid droplets. Excessive accumulation of lipids in tissues not designed for long term storage may underlie the clinical manifestations of weakened skeletal and cardiac muscle, fatty liver, pancreatitis, hypothyroidism, and type 2 diabetes.
Diagnosis
Main physical signs include a fatty liver, a weakened and enlarged heart (cardiomyopathy), inflammation of the pancreas (pancreatitis), reduced thyroid activity (hypothyroidism), type 2 diabetes, abnormal levels of creatine kinase in blood, and increased weakness of proximal muscles due to fatty replacement of skeletal muscle fibers. Accurate diagnosis is complicated by symptomatic overlap with other disorders. One of the earliest symptoms to manifest clinically is peripheral limb weakness, which becomes progressively more severe over time.[12] Specifically, asymmetric right shoulder weakness is an idiosyncratic hallmark of NLSD enabling it to be distinguished from myopathies arising from alternative muscular disorders.[13] NLSD is often diagnosed by the presence of lipid inclusions within leukocytes (Jordan’ Anomaly),[14] which can be detected using histochemical and electron microscopy.[15] Image B shows the characteristic lipid accumulation in red blood cells, in a NLSD patient, as compared to a normal individual (Image A).
Classically, NLSD is associated with myopathy and can, in some cases, be linked with ichthyosis. Therefore, with these symptoms or diagnosis, it is likely that the patient could and would be diagnosed with NLSD.[16] Although lipid accumulation is most prominent in myocytes, hepatocytes, and granulocytes, other tissues displayed elevated deposits as well. For the purpose of diagnosis, MRIs have been used to identify large fat deposits within muscle tissue.[17] People can live with NLSD, but there can be complications due to the affects this disease has on other major parts of the body like the liver, the heart, and skeletal muscle. Whereas myopathy won’t necessarily show up until a patient is in the third decade of their life,[18] a child born with ichthyosis, is immediately evaluated for NLSD, in which it is detected very early on. Also, the earlier that NLSD can be detected and symptoms treated, the better quality of life the patient can have.
Treatment
Although there is no current treatment to correct the abnormal metabolic processes underlying this disease, there are approaches to ameliorate symptoms and decrease the effects of this disease. Because there is an increase of fat storage and a decrease in fat catabolism, low fat diets are recommended for slowing the progression of the disease, including the onset of type 2 diabetes and hypothyroidism. In addition, diets containing triglycerides composed of short chain fatty acids are more beneficial than TGAs containing long chain fatty acids. Ketone bodies can be rapidly transported, catabolized, and used by many tissues including the brain. Medium-chain fatty acids in an individual’s diet are rapidly used by the body, limiting storage and therefore alleviating lipid droplet accumulation. Foods with medium-chain fatty acids include dairy, fat and coconut oil. Supplements can also be taken to increase uptake of these fatty acids. Triaheptanoin, a triglyceride containing three seven-carbon chain fatty acids, has also been proposed as a possible dietary supplement.[19] Treatment for the ichthyosis is limited; moisturizers are commonly used to help manage dry, flaky, itchy skin.
Epidemiology
The rarity and likely under-diagnosis of neutral lipid storage disease prevents an accurate epidemiological estimate of its frequency in human populations. Fewer than 100 cases of NLSD have been reported since the first case of two women with nonbullous ichthyosiform erythroderma was reported in 1974 by Maurice Dorfman.[20] Many (but not all) of the cases reported since 1974 were in individuals of Middle Eastern.descent. A possible reason for the elevated rates of occurrence may be the higher frequency of consanguineous marriages in these populations, as opposed to an elevated frequency of carriers.[21] Males and females are equally likely to be diagnosed with the disease. Genetic testing in families with a history of this disorder may be recommended.
History
Lipid inclusions in the leukocytes of blood smears were first articulated by G.H. Jordans in 1953.This is a feature of the disease, but he wasn’t necessarily finding the first case of the NLSD disease Now, having lipid deposits in the white blood cells of the individual is known and recognized as Jordan’s anomaly, due to the medical professional who discovered it.[22] The first case of Neutral Lipid Storage Disease was reported by Maurice Dorfman when he treated two sisters with non bullous ichthyosiform erythroderma in 1974.[23] His observations were then able to be confirmed with the emergence of other cases. Since then, less than 100 cases as of have been reported.[24]
See also
References
- Lefèvre C, Jobard F, Caux F, et al. (November 2001). "Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome". Am. J. Hum. Genet. 69 (5): 1002–12. doi:10.1086/324121. PMC 1274347. PMID 11590543.
- Yamaguchi, Tomohiro; Osumi, Takashi (2009). "Chanarin–Dorfman syndrome: Deficiency in CGI-58, a lipid droplet-bound coactivator of lipase". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1791 (6): 519–523. doi:10.1016/j.bbalip.2008.10.012. ISSN 1388-1981. PMID 19061969.
- Viel, Alain. "Harvard University, "Principles of Biochemistry."". YouTube. Archived from the original on 2021-12-15.
- “Neutral Lipid Storage Disease with Myopathy - Genetics Home Reference - NIH.” U.S. National Library of Medicine, National Institutes of Health. Mar 3, 2020.
- Yamaguchi, Tomohiro; Osumi, Takashi (2009). "Chanarin–Dorfman syndrome: Deficiency in CGI-58, a lipid droplet-bound coactivator of lipase". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1791
- “Neutral Lipid Storage Disease with Myopathy.” Genetic and Rare Diseases Information Center, U.S. Department of Health and Human Services. May 8, 2014
- Schweiger, Martina; Eichmann, Thomas O.; Taschler, Ulrike; Zimmermann, Robert; Zechner, Rudolf; Lass, Achim (2014-01-01), MacDougald, Ormond A. (ed.), "Chapter Ten - Measurement of Lipolysis", Methods in Enzymology, Methods of Adipose Tissue Biology, Part B, Academic Press, 538: 171–193, doi:10.1016/B978-0-12-800280-3.00010-4, PMC 4018506, PMID 24529439
- Yamaguchi, Tomohiro; Osumi, Takashi (2009). "Chanarin–Dorfman syndrome: Deficiency in CGI-58, a lipid droplet-bound coactivator of lipase". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1791 (6): 519–523. doi:10.1016/j.bbalip.2008.10.012. ISSN 1388-1981. PMID 19061969
- Missaglia, Sara; Maggi, Lorenzo; Mora, Marina; Gibertini, Sara; Blasevich, Flavia; Agostoni, Piergiuseppe; Moro, Laura; Cassandrini, Denise; Santorelli, Filippo Maria; Gerevini, Simonetta; Tavian, Daniela (May 2017). "Late onset of neutral lipid storage disease due to novel PNPLA2 mutations causing total loss of lipase activity in a patient with myopathy and slight cardiac involvement". Neuromuscular Disorders. 27 (5): 481–486. doi:10.1016/j.nmd.2017.01.011. ISSN 0960-8966. PMC 5424884. PMID 28258942.
- Yamaguchi, Tomohiro; Osumi, Takashi (2009). "Chanarin–Dorfman syndrome: Deficiency in CGI-58, a lipid droplet-bound coactivator of lipase". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1791 (6): 519–523. doi:10.1016/j.bbalip.2008.10.012. ISSN 1388-1981. PMID 19061969
- Korbelius, Melanie; Vujic, Nemanja; Sachdev, Vinay; Obrowsky, Sascha; Rainer, Silvia; Gottschalk, Benjamin; Graier, Wolfgang F.; Kratky, Dagmar (13 August 2019). "ATGL/CGI-58-Dependent Hydrolysis of a Lipid Storage Pool in Murine Enterocytes". Cell Reports. 28 (7): 1923–1934.e4. doi:10.1016/j.celrep.2019.07.030. ISSN 2211-1247. PMC 6713565. PMID 31412256.
- Zhang W, Wen B, Lu J, Zhao Y, Hong D, Zhao Z, Zhang C,i Luo Y, Qi X, Zhang Y, Song X, Zhao Y, Zhao C, Hu J, Yang H, Wang Z, Yan C, and Yuan Y. Neutral Lipid Storage Disease with Myopathy in China: A Large Multicentric Cohort Study. Orphanet J Rare Dis. Oct 2019; 14 :234.
- Zhang W, Wen B, Lu J, Zhao Y, Hong D, Zhao Z, Zhang C,i Luo Y, Qi X, Zhang Y, Song X, Zhao Y, Zhao C, Hu J, Yang H, Wang Z, Yan C, and Yuan Y. Neutral Lipid Storage Disease with Myopathy in China: A Large Multicentric Cohort Study. Orphanet J Rare Dis. Oct 2019; 14 :234.
- Zhang W, Wen B, Lu J, Zhao Y, Hong D, Zhao Z, Zhang C,i Luo Y, Qi X, Zhang Y, Song X, Zhao Y, Zhao C, Hu J, Yang H, Wang Z, Yan C, and Yuan Y. Neutral Lipid Storage Disease with Myopathy in China: A Large Multicentric Cohort Study. Orphanet J Rare Dis. Oct 2019; 14 :234.
- Yamaguchi, Tomohiro; Osumi, Takashi (2009). "Chanarin–Dorfman syndrome: Deficiency in CGI-58, a lipid droplet-bound coactivator of lipase". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1791
- Vasiljevski ER, Summers MA, Little DG, Schindeler A. Lipid storage myopathies: Current treatments and future directions. Prog Lipid Res. 2018 Oct;72:1-17. doi: 10.1016/j.plipres.2018.08.001. Epub 2018 Aug 9. Review. PubMed PMID 30099045.
- Zhang W, Wen B, Lu J, Zhao Y, Hong D, Zhao Z, Zhang C,i Luo Y, Qi X, Zhang Y, Song X, Zhao Y, Zhao C, Hu J, Yang H, Wang Z, Yan C, and Yuan Y. Neutral Lipid Storage Disease with Myopathy in China: A Large Multicentric Cohort Study. Orphanet J Rare Dis. Oct 2019; 14 :234.
- Missaglia S, Coleman RA, Mordente A, and Tavian D. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells. Feb 2019; 8(2): 187.
- Laforêt, Pascal, and Christine Vianey-Saban. "Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges." Neuromuscular Disorders 20.11 (2010): 693-700
- Yamaguchi, Tomohiro; Osumi, Takashi (2009). "Chanarin–Dorfman syndrome: Deficiency in CGI-58, a lipid droplet-bound coactivator of lipase". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1791 (6): 519–523. doi:10.1016/j.bbalip.2008.10.012. ISSN 1388-1981. PMID 19061969
- Missaglia S, Coleman RA, Mordente A, and Tavian D. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells. Feb 2019; 8(2): 187
- Vasiljevski ER, Summers MA, Little DG, Schindeler A. Lipid storage myopathies: Current treatments and future directions. Prog Lipid Res. 2018 Oct;72:1-17. doi: 10.1016/j.plipres.2018.08.001. Epub 2018 Aug 9. Review. PubMed PMID 30099045.
- Yamaguchi, Tomohiro; Osumi, Takashi (2009). "Chanarin–Dorfman syndrome: Deficiency in CGI-58, a lipid droplet-bound coactivator of lipase". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1791 (6): 519–523. doi:10.1016/j.bbalip.2008.10.012. ISSN 1388-1981. PMID 19061969
- Winokur, et al. “Jordans Anomaly.” Blood, American Society of Hematology, 15 Feb. 2018