G protein-gated ion channel
G protein-gated ion channels are a family of transmembrane ion channels in neurons and atrial myocytes that are directly gated by G proteins.
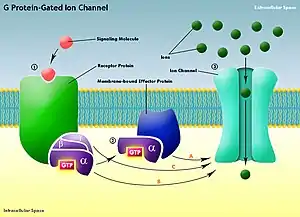
Overview of mechanisms and function
Generally, G protein-gated ion channels are specific ion channels located in the plasma membrane of cells that are directly activated by a family of associated proteins. Ion channels allow for the selective movement of certain ions across the plasma membrane in cells. More specifically, in nerve cells, along with ion transporters, they are responsible for maintaining the electrochemical gradient across the cell.
G proteins are a family of intracellular proteins capable of mediating signal transduction pathways. Each G protein is a heterotrimer of three subunits: α-, β-, and γ- subunits. The α-subunit (Gα) typically binds the G protein to a transmembrane receptor protein known as a G protein-coupled receptor, or GPCR. This receptor protein has a large, extracellular binding domain which will bind its respective ligands (e.g. neurotransmitters and hormones). Once the ligand is bound to its receptor, a conformational change occurs. This conformational change in the G protein allows Gα to bind GTP. This leads to yet another conformational change in the G protein, resulting in the separation of the βγ-complex (Gβγ) from Gα.[1] At this point, both Gα and Gβγ are active and able to continue the signal transduction pathway. Different classes of G protein-coupled receptors have many known functions including the cAMP and Phosphatidylinositol signal transduction pathways.[2] A class known as metabotropic glutamate receptors play a large role in indirect ion channel activation by G proteins. These pathways are activated by second messengers which initiate signal cascades involving various proteins which are important to the cell's response.
G protein-gated ion channels are associated with a specific type of G protein-coupled receptor. These ion channels are transmembrane ion channels with selectivity filters and a G protein binding site. The GPCRs associated with G protein-gated ion channels are not involved in signal transduction pathways. They only directly activate these ion channels using effector proteins or the G protein subunits themselves (see picture). Unlike most effectors, not all G protein-gated ion channels have their activity mediated by Gα of their corresponding G proteins. For instance, the opening of inwardly rectifying K+ (GIRK) channels is mediated by the binding of Gβγ.[3]
G protein-gated ion channels are primarily found in CNS neurons and atrial myocytes, and affect the flow of potassium (K+), calcium (Ca2+), sodium (Na+), and chloride (Cl−) across the plasma membrane.[4]
Types of G Protein-gated ion channels
Potassium channels
Structure
Four G protein gated inwardly-rectifying potassium (GIRK) channel subunits have been identified in mammals: GIRK1, GIRK2, GIRK3, and GIRK4. The GIRK subunits come together to form GIRK ion channels. These ion channels, once activated, allow for the flow of potassium ions (K+) from the extracellular space surrounding the cell across the plasma membrane and into the cytoplasm. Each channel consists of domains which span the plasma membrane, forming the K+-selective pore region through which the K+ ions will flow.[5][6] Both the N-and C-terminal ends of the GIRK channels are located within the cytoplasm. These domains interact directly with the βγ-complex of the G protein, leading to activation of the K+ channel. .[7] These domains on the N-and C-terminal ends which interact with the G proteins contain certain residues which are critical for the proper activation of the GIRK channel. In GIRK4, the N-terminal residue is His-64 and the C-terminal residue is Leu-268; in GIRK1 they are His-57 and Leu-262, respectively. Mutations in these domains lead to the channel's desensitivity to the βγ-complex and therefore reduce the activation of the GIRK channel.[3]
The four GIRK subunits are 80-90% similar in their pore-forming and transmembrane domains, a feature accountable by the similarities in their structures and sequences. GIRK2, GIRK3, and GIRK4 share an overall identity of 62% with each other, while GIRK1 only shares 44% identity with the others.[6] Because of their similarity, the GIRK channel subunits can come together easily to form heteromultimers (a protein with two or more different polypeptide chains). GIRK1, GIRK2, and GIRK3 show abundant and overlapping distribution in the central nervous system (CNS) while GIRK1 and GIRK4 are found primarily in the heart.[4] GIRK1 combines with GIRK2 in the CNS and GIRK4 in the atrium to form heterotetramers; each final heterotetramer contains two GIRK1 subunits and two GIRK2 or GIRK4 subunits. GIRK2 subunits can also form homotetramers in the brain, while GIRK4 subunits can form homotetramers in the heart.[7] GIRK1 subunits have not been shown to be able to form functional homotetramers. Though GIRK3 subunits are found in the CNS, their role in forming functional ion channels is still unknown.[4]
Subtypes and respective functions
- GIRKs found in the heart
One G protein-gated potassium channel is the inward-rectifing potassium channel (IKACh) found in cardiac muscle (specifically, the sinoatrial node and atria),[8] which contributes to the regulation of heart rate.[9] These channels are almost entirely dependent on G protein activation, making them unique when compared to other G protein-gated channels.[10] Activation of the IKACh channels begins with release of acetylcholine (ACh) from the vagus nerve[9] onto pacemaker cells in the heart.[10] ACh binds to the M2 muscarinic acetylcholine receptors, which interact with G proteins and promote the dissociation of the Gα subunit and Gβγ-complex.[11] IKACh is composed of two homologous GIRK channel subunits: GIRK1 and GIRK4. The Gβγ-complex binds directly and specifically to the IKACh channel through interactions with both the GIRK1 and GIRK4 subunits.[12] Once the ion channel is activated, K+ ions flow out of the cell and cause it to hyperpolarize.[13] In its hyperpolarized state, the neuron cannot fire action potentials as quickly, which slows the heartbeat.[14]
- GIRKs found in the brain
The G protein inward rectifying K+ channel found in the CNS is a heterotetramer composed of GIRK1 and GIRK2 subunits[4] and is responsible for maintaining the resting membrane potential and excitability of the neuron.[9] Studies have shown the largest concentrations of the GIRK1 and GIRK2 subunits to be in the dendritic areas of neurons in the CNS.[4] These areas, which are both extrasynaptic (exterior to a synapse) and perisynaptic (near a synapse), correlate with the large concentration of GABAB receptors in the same areas. Once the GABAB receptors are activated by their ligands, they allow for the dissociation of the G protein into its individual α-subunit and βγ-complex so it can in turn activate the K+ channels. The G proteins couple the inward rectifying K+ channels to the GABAB receptors, mediating a significant part of the GABA postsynaptic inhibition.[4]
Furthermore, GIRKs have been found to play a role in a group of serotonergic neurons in the dorsal raphe nucleus, specifically those associated with the neuropeptide hormone orexin.[15] The 5-HT1A receptor, a serotonin receptor and type of GPCR, has been shown to be coupled directly with the α-subunit of a G protein, while the βγ-complex activates GIRK without use of a second messenger. The subsequent activation of the GIRK channel mediates hyperpolarization of orexin neurons, which regulate the release of many other neurotransmitters including noradrenaline and acetylcholine.[15]
Structure
In addition to the subset of potassium channels that are directly gated by G proteins, G proteins can also directly gate certain calcium ion channels in neuronal cell membranes. Although membrane ion channels and protein phosphorylation are typically indirectly affected by G protein-coupled receptors via effector proteins (such as phospholipase C and adenylyl cyclase) and second messengers (such as inositol triphosphate, diacylglycerol and cyclic AMP), G proteins can short circuit the second-messenger pathway and gate the ion channels directly.[16] Such bypassing of the second-messenger pathways is observed in mammalian cardiac myocytes and associated sarcolemmal vesicles in which Ca2+ channels are able to survive and function in the absence of cAMP, ATP or protein kinase C when in the presence of the activated α-subunit of the G protein.[17] For example, Gα, which is stimulatory to adenylyl cyclase, acts on the Ca2+ channel directly as an effector. This short circuit is membrane-delimiting, allowing direct gating of calcium channels by G proteins to produce effects more quickly than the cAMP cascade could.[16] This direct gating has also been found in specific Ca2+ channels in the heart and skeletal muscle T tubules.[18]
Function
Several high-threshold, slowly inactivating calcium channels in neurons are regulated by G proteins.[13] The activation of α-subunits of G proteins has been shown to cause rapid closing of voltage-dependent Ca2+ channels, which causes difficulties in the firing of action potentials.[1] This inhibition of voltage-gated Calcium channels by G protein-coupled receptors has been demonstrated in the dorsal root ganglion of a chick among other cell lines.[13] Further studies have indicated roles for both Gα and Gβγ subunits in the inhibition of Ca2+ channels. The research geared to defining the involvement of each subunit, however, has not uncovered the specificity or mechanisms by which Ca2+ channels are regulated.
The acid-sensing ion channel ASIC1a is a specific G protein-gated Ca2+ channel. The upstream M1 muscarinic acetylcholine receptor binds to Gq-class G proteins. Blocking this channel with the agonist oxotremorine methiodide was shown to inhibit ASIC1a currents.[19] ASIC1a currents have also been shown to be inhibited in the presence of oxidizing agents and potentiated in the presence of reducing agents. A decrease and increase in acid-induced intracellular Ca2+ accumulation were found, respectively.[20]
Sodium channels
Patch clamp measurements suggest a direct role for Gα in the inhibition of fast Na+ current within cardiac cells.[21] Other studies have found evidence for a second-messenger pathway which may indirectly control these channels.[22] Whether G proteins indirectly or directly activate Na+ ion channels not been defined with complete certainty.
Chloride channels
Chloride channel activity in epithelial and cardiac cells has been found to be G protein-dependent. However, the cardiac channel that has been shown to be directly gated by the Gα subunit has not yet been identified. As with Na+ channel inhibition, second-messenger pathways cannot be discounted in Cl− channel activation.[23]
Studies done on specific Cl− channels show differing roles of G protein activation. It has been shown that G proteins directly activate one type of Cl− channel in skeletal muscle.[10] Other studies, in CHO cells, have demonstrated a large conductance Cl− channel to be activated differentially by CTX- and PTX-sensitive G proteins.[10] The role of G proteins in the activation of Cl− channels is a complex area of research that is ongoing.
Clinical significance and ongoing research
Mutations in G proteins associated with G protein-gated ion channels have been shown to be involved in diseases such as epilepsy, muscular diseases, neurological diseases, and chronic pain, among others.[4]
Epilepsy, chronic pain, and addictive drugs such as cocaine, opioids, cannabinoids, and ethanol all affect neuronal excitability and heart rate. GIRK channels have been shown to be involved in seizure susceptibility, cocaine addiction, and increased tolerance for pain by opioids, cannabinoids, and ethanol.[24] This connection suggests that GIRK channel modulators may be useful therapeutic agents in the treatment of these conditions. GIRK channel inhibitors may serve to treat addictions to cocaine, opioids, cannabinoids, and ethanol while GIRK channel activators may serve to treat withdrawal symptoms.[24]
Alcohol intoxication
Alcohol intoxication has been shown to be directly connected to the actions of GIRK channels. GIRK channels have a hydrophobic pocket that is capable of binding ethanol, the type of alcohol found in alcoholic beverages.[25][26] When ethanol acts as an agonist, GIRK channels in the brain experience prolonged opening. This causes decreased neuronal activity, the result of which manifests as the symptoms of alcohol intoxication. The discovery of the hydrophobic pocket capable of binding ethanol is significant in the field of clinical pharmacology. Agents that can act as agonists to this binding site can be potentially useful in the creation of drugs for the treatment of neurological disorders such as epilepsy in which neuronal firing exceeds normal levels.[26]
Breast cancer
Studies have shown that a link exists between channels with GIRK1 subunits and the beta-adrenergic receptor pathway in breast cancer cells responsible for growth regulation of the cells. Approximately 40% of primary human breast cancer tissues have been found to carry the mRNA which codes for GIRK1 subunits.[27] Treatment of breast cancer tissue with alcohol has been shown to trigger increased growth of the cancer cells. The mechanism of this activity is still a subject of research.[27]
Down syndrome
Altered cardiac regulation is common in adults diagnosed with Down syndrome and may be related to G protein-gated ion channels. The KCNJ6 gene is located on chromosome 21 and encodes for the GIRK2 protein subunit of G protein-gated K+ channels.[28] People with Down Syndrome have three copies of chromosome 21, resulting in an overexpression of the GIRK2 subunit. Studies have found that recombinant mice overexpressing GIRK2 subunits show altered responses to drugs that activate G protein-gated K+ channels. These altered responses were limited to the sino-atrial node and atria, both areas which contain many G protein-gated K+ channels.[28] Such findings could potentially lead to the development of drugs that can help regulate the cardiac sympathetic-parasympathetic imbalance in Down Syndrome adults.
Chronic atrial fibrillation
Atrial fibrillation (abnormal heart rhythm) is associated with shorter action potential duration and believed to be affected by the G protein-gated K+ channel, IK,ACh.[29] The IK,ACh channel, when activated by G proteins, allows for the flow of K+ across the plasma membrane and out of the cell. This current hyperpolarizes the cell, thus terminating the action potential. It has been shown that in chronic atrial fibrillation there an increase in this inwardly rectifying current because of constantly activated IK,ACh channels.[29] Increase in the current results in shorter action potential duration experienced in chronic atrial fibrillation and leads to the subsequent fibrillating of the cardiac muscle. Blocking IK,ACh channel activity could be a therapeutic target in atrial fibrillation and is an area under study.
Pain management
GIRK channels have been demonstrated in vivo to be involved in opioid- and ethanol-induced analgesia.[30] These specific channels have been the target of recent studies dealing with genetic variance and sensitivity to opioid analgesics due to their role in opioid-induced analgesia. Several studies have shown that when opioids are prescribed to treat chronic pain, GIRK channels are activated by certain GPCRs, namely opioid receptors, which leads to the inhibition of nociceptive transmission, thus functioning in pain relief.[31] Furthermore, studies have shown that G proteins, specifically the Gi alpha subunit, directly activate GIRKs which were found to participate in propagation of morphine-induced analgesia in inflamed spines of mice.[32] Research pertaining to chronic pain management continues to be performed in this field.
See also
- G protein
- G protein-coupled receptor
- Metabotropic receptor
References
- Stryer L, Berg JM, Tymoczko JL (2007). Biochemistry (6th ed.). San Francisco: W.H. Freeman. ISBN 978-0-7167-8724-2.
- Gilman AG (1987). "G Proteins: Transducers of Receptor-Generated Signals". Annual Review of Biochemistry. 56: 615–49. doi:10.1146/annurev.bi.56.070187.003151. PMID 3113327.
- He C, Yan X, Zhang H, Mirshahi T, Jin T, Huang A, Logothetis DE (February 2002). "Identification of critical residues controlling G protein-gated inwardly rectifying K(+) channel activity through interactions with the beta gamma subunits of G proteins". The Journal of Biological Chemistry. 277 (8): 6088–96. doi:10.1074/jbc.M104851200. PMID 11741896.
- Koyrakh L, Luján R, Colón J, Karschin C, Kurachi Y, Karschin A, Wickman K (December 2005). "Molecular and cellular diversity of neuronal G-protein-gated potassium channels". The Journal of Neuroscience. 25 (49): 11468–78. doi:10.1523/JNEUROSCI.3484-05.2005. PMC 6725904. PMID 16339040.
- Neer EJ, Clapham DE (Jan–Feb 1992). "Signal transduction through G proteins in the cardiac myocyte". Trends in Cardiovascular Medicine. 2 (1): 6–11. doi:10.1016/1050-1738(92)90037-S. PMID 21239281.
- Jelacic TM, Sims SM, Clapham DE (May 1999). "Functional expression and characterization of G-protein-gated inwardly rectifying K+ channels containing GIRK3". The Journal of Membrane Biology. 169 (2): 123–9. doi:10.1007/s002329900524. PMID 10341034. S2CID 13538678.
- Yakubovich D, Pastushenko V, Bitler A, Dessauer CW, Dascal N (May 2000). "Slow modal gating of single G protein-activated K+ channels expressed in Xenopus oocytes". The Journal of Physiology. 524 (Pt 3): 737–55. doi:10.1111/j.1469-7793.2000.00737.x. PMC 2269908. PMID 10790155.
- Nikolov EN, Ivanova-Nikolova TT (May 2004). "Coordination of membrane excitability through a GIRK1 signaling complex in the atria". The Journal of Biological Chemistry. 279 (22): 23630–6. doi:10.1074/jbc.M312861200. PMID 15037627.
- Mark MD, Herlitze S (October 2000). "G-protein mediated gating of inward-rectifier K+ channels". European Journal of Biochemistry. 267 (19): 5830–6. doi:10.1046/j.1432-1327.2000.01670.x. PMID 10998041.
- Wickman KD, Clapham DE (June 1995). "G-protein regulation of ion channels". Current Opinion in Neurobiology. 5 (3): 278–85. doi:10.1016/0959-4388(95)80039-5. PMID 7580149. S2CID 27125330.
- Ivanova-Nikolova TT, Nikolov EN, Hansen C, Robishaw JD (August 1998). "Muscarinic K+ channel in the heart. Modal regulation by G protein beta gamma subunits". The Journal of General Physiology. 112 (2): 199–210. doi:10.1085/jgp.112.2.199. PMC 2525744. PMID 9689027.
- Corey S, Krapivinsky G, Krapivinsky L, Clapham DE (February 1998). "Number and stoichiometry of subunits in the native atrial G-protein-gated K+ channel, IKACh". The Journal of Biological Chemistry. 273 (9): 5271–8. doi:10.1074/jbc.273.9.5271. PMID 9478984.
- Morris AJ, Malbon CC (October 1999). "Physiological regulation of G protein-linked signaling". Physiological Reviews. 79 (4): 1373–430. doi:10.1152/physrev.1999.79.4.1373. PMID 10508237. S2CID 26873265.
- Fitzpatrick D, Purves D, Augustine G (2004). Neuroscience. Sunderland, Mass: Sinauer. ISBN 978-0-87893-725-7.
- Nishino S, Sakuri T, eds. (2006). The Orexin/Hypocretin System. Totowa, NJ: Humana Press. ISBN 978-1-58829-444-9.
- Brown AM, Yatani A, Imoto Y, Kirsch G, Hamm H, Codina J, et al. (1988). "Direct coupling of G proteins to ionic channels". Cold Spring Harbor Symposia on Quantitative Biology. 53 (1): 365–73. doi:10.1101/sqb.1988.053.01.044. PMID 3151174.
- Yatani A, Codina J, Imoto Y, Reeves JP, Birnbaumer L, Brown AM (November 1987). "A G protein directly regulates mammalian cardiac calcium channels". Science. 238 (4831): 1288–92. Bibcode:1987Sci...238.1288Y. doi:10.1126/science.2446390. PMID 2446390.
- Brown AM, Birnbaumer L (March 1988). "Direct G protein gating of ion channels". The American Journal of Physiology. 254 (3 Pt 2): H401-10. doi:10.1152/ajpheart.1988.254.3.H401. PMID 2450476.
- Dorofeeva NA, Karpushev AV, Nikolaev MV, Bolshakov KV, Stockand JD, Staruschenko A (October 2009). "Muscarinic M1 modulation of acid-sensing ion channels". NeuroReport. 20 (15): 1386–91. doi:10.1097/WNR.0b013e3283318912. PMID 19730136. S2CID 36155539.
- Chu XP, Close N, Saugstad JA, Xiong ZG (May 2006). "ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents". The Journal of Neuroscience. 26 (20): 5329–39. doi:10.1523/JNEUROSCI.0938-06.2006. PMC 3799800. PMID 16707785.
- Schubert B, VanDongen AM, Kirsch GE, Brown AM (August 1989). "Beta-adrenergic inhibition of cardiac sodium channels by dual G-protein pathways". Science. 245 (4917): 516–9. Bibcode:1989Sci...245..516S. doi:10.1126/science.2547248. PMID 2547248.
- Ling BN, Kemendy AE, Kokko KE, Hinton CF, Marunaka Y, Eaton DC (December 1990). "Regulation of the amiloride-blockable sodium channel from epithelial tissue". Molecular and Cellular Biochemistry. 99 (2): 141–50. doi:10.1007/BF00230344. PMID 1962846. S2CID 24533531.
- Fargon F, McNaughton PA, Sepúlveda FV (October 1990). "Possible involvement of GTP-binding proteins in the deactivation of an inwardly rectifying K+ current in enterocytes isolated from guinea-pig small intestine". Pflügers Archiv. 417 (2): 240–2. doi:10.1007/BF00370706. PMID 1707517. S2CID 8807951.
- Kobayashi T, Washiyama K, Ikeda K (October 2004). "Modulators of G protein-activated inwardly rectifying K+ channels: potentially therapeutic agents for addictive drug users". Annals of the New York Academy of Sciences. 1025 (1): 590–4. Bibcode:2004NYASA1025..590K. doi:10.1196/annals.1316.073. PMID 15542767. S2CID 26047083.
- Aryal P, Dvir H, Choe S, Slesinger PA (August 2009). "A Discrete Alcohol Pocket Involved in GIRK Channel Activation". Nature Neuroscience. 12 (8): 988–95. doi:10.1038/nn.2358. PMC 2717173. PMID 19561601.
- Lewohl JM, Wilson WR, Mayfield RD, Brozowski SJ, Morrisett RA, Harris RA (December 1999). "G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action" (PDF). Nature Neuroscience. 2 (12): 1084–90. doi:10.1038/16012. PMID 10570485. S2CID 292545. Archived from the original (PDF) on 2005-01-23.
- Dhar MS, Plummer HK (August 2006). "Protein expression of G-protein inwardly rectifying potassium channels (GIRK) in breast cancer cells". BMC Physiology. 6: 8. doi:10.1186/1472-6793-6-8. PMC 1574343. PMID 16945134.
- Lignon JM, Bichler Z, Hivert B, Gannier FE, Cosnay P, del Rio JA, et al. (April 2008). "Altered heart rate control in transgenic mice carrying the KCNJ6 gene of the human chromosome 21". Physiological Genomics. 33 (2): 230–9. doi:10.1152/physiolgenomics.00143.2007. PMID 18303085.
- Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, et al. (December 2005). "The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation". Circulation. 112 (24): 3697–706. doi:10.1161/CIRCULATIONAHA.105.575332. PMID 16330682.
- Marker CL, Luján R, Loh HH, Wickman K (April 2005). "Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids". The Journal of Neuroscience. 25 (14): 3551–9. doi:10.1523/JNEUROSCI.4899-04.2005. PMC 6725379. PMID 15814785.
- Nishizawa D, Nagashima M, Katoh R, Satoh Y, Tagami M, Kasai S, et al. (September 2009). Zanger U (ed.). "Association between KCNJ6 (GIRK2) Gene Polymorphisms and Postoperative Analgesic Requirements after Major Abdominal Surgery". PLOS ONE. 4 (9): e7060. Bibcode:2009PLoSO...4.7060N. doi:10.1371/journal.pone.0007060. PMC 2738941. PMID 19756153.
- González-Rodríguez S, Hidalgo A, Baamonde A, Menéndez L (January 2010). "Involvement of Gi/o proteins and GIRK channels in the potentiation of morphine-induced spinal analgesia in acutely inflamed mice". Naunyn-Schmiedeberg's Archives of Pharmacology. 381 (1): 59–71. doi:10.1007/s00210-009-0471-3. PMID 19940980. S2CID 10134890.