Hemoglobin M disease
Hemoglobin M disease is a rare form of hemoglobinopathy, characterized by the presence of hemoglobin M (HbM) and elevated methemoglobin (metHb) level in blood.[1] HbM is an altered form of hemoglobin (Hb) due to point mutation occurring in globin-encoding genes, mostly involving tyrosine substitution for proximal (F8) or distal (E7) histidine residues.[2] HbM variants are inherited as autosomal dominant disorders and have altered oxygen affinity.[3] The pathophysiology of hemoglobin M disease involves heme iron autoxidation promoted by heme pocket structural alteration.[4]
| Hemoglobin M disease | |
|---|---|
 | |
| Bluish fingertips in a cyanotic patient | |
| Symptoms | Cyanosis and dark brown blood |
| Causes | Hemoglobin M variants |
| Diagnostic method | Hemoglobin electrophoresis, UV spectroscopy, DNA sequencing, etc. |
| Treatment | No treatment is required |
There exists at least 13 HbM variants, such as Boston, Osaka, Saskatoon, etc., named according to their geographical locations of discovery. Different HbM variants may give different signs and symptoms. Major signs include cyanosis and dark brown blood. Patients may be asymptomatic or experience dizziness, headache, mild dyspnea, etc.[2][5] Diagnosis is usually suspected based on cyanosis. Biochemical testing, hemoglobin electrophoresis, ultraviolet-visible wavelength light spectroscopy, and DNA-based globin gene analysis can be used for diagnosis.[2][5][6][7][8][9][10] Hemoglobin M disease is often not life-threatening and there is no known effective treatment.[3][5][10][11][12][13]
Hemoglobin M disease is a congenital subtype of methemoglobinemia.[2] For other congenital subtypes of methemoglobinemia, cytochrome b5 reductase (CYB5R) deficiency is the major cause, rendering defective conversion of metHb to normal Hb. CYB5R deficiency is an autosomal recessive condition.[14]
.jpg.webp)
Signs and Symptoms
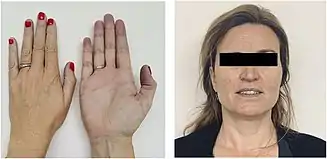
Cyanosis is the most common sign of hemoglobin M disease, which can be observed in all kinds of hemoglobin M diseases. It is mostly presented in the patient's lips and fingertips.[15] Cyanosis in hemoglobin M disease results from elevated levels of metHb and sulfhemoglobin (sulfHb).[16] Dark brown blood is another major sign of hemoglobin M disease. Hemoglobin M diseases caused by different HbM variants may have slight variations in their signs and symptoms, some include signs such as hemolytic anemia, decreased HbA1c, and abnormal co-oximetry.[13][17][18] Onset of cyanosis varies among alpha-, beta- and gamma-chain variants. Infants with alpha- or gamma-chain variants manifest cyanosis since birth but transient neonatal cyanosis caused by gamma-chain variants resolves soon after the disappearance of fetal Hb. Infants with beta-globin variants become cyanotic around 6 months after birth with the completion of the fetal-to-adult Hb switch.[19]
Hemoglobin M disease is usually asymptomatic. However, it may show symptoms such as confusion, headache, tachycardia, and mild dyspnea at metHb level ranging from 10% to 30%. Other possible moderate signs and symptoms (metHb level above 30%) include dizziness, syncope, chest pain, palpitations, and fatigue. More severe signs (metHb level above 50%) include tachypnea, metabolic acidosis, dysrhythmia, seizure, delirium, coma, and death (metHb level above 70%).[5]
Pathophysiology
HbM is a rare methemoglobin group inherited in an autosomal dominant manner, resulting from missense mutations in genes encoding alpha (HBA1, HBA2), beta (HBB), or gamma (HBG1, HBG2) globin chains. In most HbM variants, the proximal (F8) or distal (E7) histidine residue is replaced by tyrosine.[2][3] Proximal histidine (F8) is designated as position 87 in the alpha chain and 92 in the beta chain. Distal histidine (E7) is designated as position 58 in the alpha chain and position 63 in the beta chain.[5]
Different HbM Variants
At least 13 HbM variants involving alpha- or beta- or gamma-chains have been reported. Six variants, namely HbM Boston, HbM Iwate, HbM Saskatoon, HbM Hyde Park, HbFM Osaka and HbFM Fort Ripley, manifest proximal (F8) or distal (E7) histidine substitution by tyrosine at position alpha-58, alpha-87, beta-63, beta-92, gamma-63 and gamma-92 respectively. HbM Milwaukee-1 involves valine (E11) substitution by glutamate residue at position beta-67.[10]
Alterations in Oxygen Affinity
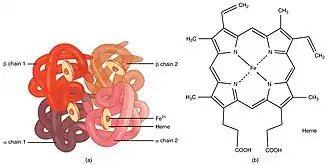
Under normal circumstances, the heme iron in ferrous state (Fe2+) is covalently bound to imidazole nitrogen of the proximal histidine (F8), and is able to bind to an oxygen molecule.[5]
Tyrosine substitution renders structural alteration of the heme pocket and promotes spontaneous oxidation of heme iron from its ferrous state to ferric state (Fe3+) through discharge of a superoxide ion. Tyrosine can form an iron-phenolate complex with ferric iron which prevents reduction back to divalent ferrous iron.[5][20] Stable covalent bond between tyrosine and Fe3+ hinders interaction between ferric iron and oxygen. Inability of ferric heme iron in binding oxygen alters oxygen affinity of ferrous heme iron in the remaining normal subunits, impeding oxygen delivery to body tissues.[8][21][22]
Stabilization of ferric iron is done through various abnormal coordination mechanisms between mutant side chains and ferric iron.[22]
Mechanisms of Different Hemoglobin M Diseases
Deoxygenated T (Tensed) state (low-affinity Hb quaternary structure)[23] is stabilized in alpha-chain variants due to constraints between subunits. Ferric iron in alpha-chain variants show exceptional resistance to enzymatic reduction by metHb reductases or chemical reduction. In HbM Boston (alpha-58 [E7] His→Tyr), new Tyr (E7) coordination alters the heme plane to disrupt the normal interaction between proximal His (F8) and heme iron. In HbM Iwate (alpha-87 [F8] His→Tyr), tyrosine coordination distorts the heme position. This increases the separation between heme group and helix F within the altered alpha subunits for ferric iron stabilization.[22][24]
Oxygenated R (Relaxed) state (high-affinity Hb quaternary structure)[23] is stabilized in beta-globin variants due to relaxed constraints between subunits. In HbM Saskatoon (beta-63 [E7] His→Tyr), substitution of a larger Tyr (E7) renders close proximity with heme ferric iron hence forming a hexacoordinate iron site where transient protonation of Tyr (E7) prompts enzymatic reduction by metHb reductase.[13][22][24] In HbM Hyde Park (beta-92 [F8] His→Tyr), heme loss and instability with nearby residue reconstruction contribute to its pathophysiology.[22][24][25]
Physiological properties of non-mutant subunits within the mutant Hb tetramer differ. Normal alpha subunits in beta-chain variants (HbM Saskatoon and HbM Hyde Park) exhibit significant cooperativity and Bohr effect, displaying an increased oxygen affinity. Normal beta subunits in alpha-chain variants (HbM Boston and HbM Iwate) exhibit reduced cooperativity and Bohr effect, displaying a decreased oxygen affinity. Hence, lower circulating oxidized Hb is observed in beta-chain variants than that in alpha-chain variants.[22][24][25]
For HbM Milwaukee-1 (beta-67 [E11] Val→Glu), proximity of anionic glutamate to the heme iron favors the autoxidation of ferrous iron and stabilization of ferric iron by direct coordination to its sixth coordinate position. This decreases oxygen affinity.[22][26]
Diagnosis
Cyanosis caused by hemoglobin M disease is often mistaken as cardiac or pulmonary defects. Correct diagnosis is important to prevent unnecessary invasive procedures such as cardiac catheterization and mechanical ventilation.[5][22]
Biochemical Testing
Exposure of venous blood samples to pure oxygen can be used to differentiate cyanosis caused by metHb from cardiopulmonary cyanosis or other cyanosis caused by low-O2 affinity Hbs. Cyanotic patients with methemoglobinemia display brownish blood while purple deoxyhemoglobin becomes bright red oxyhemoglobin in other cases.[10] Addition of potassium cyanide (KCN) can be used to further distinguish hemoglobin M disease from other subtypes of methemoglobinemia and sulfhemoglobinemia. For sulfhemoglobinemia, sulfHb is inert to cyanide and shows no colour change. Hemolysates containing metHb with wild-type globin chains turn red immediately. The color change in hemolysates containing metHb with mutated globin chains is slower and the conversion rate for different HbM variants may vary.[22]
Hemoglobin Electrophoresis
.jpg.webp)
It provides qualitative analysis by identification of abnormal Hb variants. Addition of KCN before electrophoresis converts all Hb types into metHb to prevent result misinterpretation due to iron state differences. Normal and abnormal Hb variants are separated by electric current, and the observed differences in migration indicate the substitution of the amino acid.[22] For clear separation, hemoglobin electrophoresis should be performed on agar gel at pH 7.1. Under alkaline conditions, HbM migrates slightly slower than HbA.[3] Further confirmatory testing can be performed by high-performance liquid chromatography (HPLC) to provide quantification of the Hb fractions.[6][7][8]
Ultraviolet-Visible Wavelength Light Spectroscopy
Spectral absorption of the hemolysate at various wavelengths can be used for diagnosis.[3] Compared with normal blood, a unique absorption range of HbM variants can be seen. HbM exhibits a specific light absorption pattern, shown by visible peaks at 510 and 630 nm. This explains the formation of chocolate-brown blood.[6] CO-oximetry using multiple wavelengths is preferred over pulse oximetry in metHb detection. Pulse oximetry only uses two distinct wavelengths of 660 and 940 nm which can be misleading.[12][13]
Treatment
Hemoglobin M disease is often not life-threatening and treatment is not necessary. There is no existing effective treatment, including methylene blue (MB) and ascorbic acid used in treating acquired methemoglobinemia.[12] MB is an oxidant and it is not used to treat hemoglobin M disease. They are prone to develop symptomatic methemoglobinemia given further exposure to oxidants.[13]
References
- "Hemoglobin M disease (Concept Id: C3665425) – MedGen – NCBI". www.ncbi.nlm.nih.gov. Retrieved 2022-03-28.
- Ludlow, John T.; Wilkerson, Richard G.; Nappe, Thomas M. (2022), "Methemoglobinemia", StatPearls, Treasure Island (FL): StatPearls Publishing, PMID 30726002, retrieved 2022-03-28
- Randolph, Tim R. (2020-01-01), Keohane, Elaine M.; Otto, Catherine N.; Walenga, Jeanine M. (eds.), "24 - Hemoglobinopathies (structural defects in hemoglobin)", Rodak's Hematology (Sixth Edition), St. Louis (MO): Elsevier, pp. 394–423, ISBN 978-0-323-53045-3, retrieved 2022-03-28
- Alonso-Ojembarrena, A. (2016). "Hemoglobin M Disease as a Cause of Cyanosis in a Newborn". Journal of Pediatric Hematology/Oncology. 38 (3): 173–175. doi:10.1097/MPH.0000000000000489. PMID 26694193. S2CID 26848356 – via PubMed.
- Iolascon, Achille; Bianchi, Paola; Andolfo, Immacolata; Russo, Roberta; Barcellini, Wilma; Fermo, Elisa; Toldi, Gergely; Ghirardello, Stefano; Rees, Davis; Van Wijk, Richard; Kattamis, Antonis (2021-09-23). "Recommendations for diagnosis and treatment of methemoglobinemia". American Journal of Hematology. 96 (12): 1666–1678. doi:10.1002/ajh.26340. ISSN 0361-8609. PMID 34467556. S2CID 237377613.
- Picca, Andrew; Ruthford, Mason; Ghanim, Majd T.; Sims, Morgan; Kanter, Julie (2019-08-20). "Diagnosis of Hemoglobin M Disease in a Toddler Presenting With Hypoxemia and Hemolysis". Clinical Pediatrics. 58 (11–12): 1345–1348. doi:10.1177/0009922819870555. ISSN 0009-9228. PMID 31431070. S2CID 201115221.
- Leung, Kwok Yin; Au, Patrick; Tang, Mary (2020-01-01), Pandya, Pranav P.; Oepkes, Dick; Sebire, Neil J.; Wapner, Ronald J. (eds.), "27 - Prenatal Screening for Thalassemias", Fetal Medicine (Third Edition), London: Elsevier, pp. 263–273.e1, ISBN 978-0-7020-6956-7, retrieved 2022-03-28
- Arceci, Robert J.; Hann, Ian M.; Smith, Owen P., eds. (2006-08-31). Pediatric Hematology. Blackwell Publishing. doi:10.1002/9780470987001. ISBN 9780470987001.
- Sangkitporn, S. K.; Eksiri, L.; Sangnoi, A.; Duangruang, S.; Dumbua, A.; Rattanakittisophon, K.; Sangkitporn, S. (2009). "Identification of beta-globin gene mutations in Thailand using an automated fluorescence-based DNA sequencer". International Journal of Laboratory Hematology. 31 (5): 521–527. doi:10.1111/j.1751-553X.2008.01072.x. ISSN 1751-553X. PMID 18498386. S2CID 31589532.
- Old, John (2013-01-01), Rimoin, David; Pyeritz, Reed; Korf, Bruce (eds.), "Chapter 71 - Hemoglobinopathies and Thalassemias", Emery and Rimoin's Principles and Practice of Medical Genetics (Sixth Edition), Oxford: Academic Press, pp. 1–44, ISBN 978-0-12-383834-6, retrieved 2022-03-28
- Rehman, Habib Ur (2001). "Methemoglobinemia". Western Journal of Medicine. 175 (3): 193–196. doi:10.1136/ewjm.175.3.193. ISSN 0093-0415. PMC 1071541. PMID 11527852.
- Patnaik, Sibabratta; Natarajan, Manivachagan Muthappa; James, Ebor Jacob; Ebenezer, Kala (2014). "Methylene blue unresponsive methemoglobinemia". Indian Journal of Critical Care Medicine. 18 (4): 253–255. doi:10.4103/0972-5229.130582. ISSN 0972-5229. PMC 4033863. PMID 24872659.
- Göttgens, Eva-Leonne; Baks, Kristian; Harteveld, Cornelis L.; Goossens, Kristel; van Gammeren, Adriaan J. (2021). "Cyanosis, hemolysis, decreased HbA1c and abnormal co-oximetry in a patient with hemoglobin M Saskatoon [HBB:c.190C > T p.His64Tyr]". Hematology (Amsterdam, Netherlands). 26 (1): 914–918. doi:10.1080/16078454.2021.1999048. ISSN 1607-8454. PMID 34789072. S2CID 244346360.
- Percy, Melanie J.; Lappin, Terry R. (2008). "Recessive congenital methaemoglobinaemia: cytochrome b(5) reductase deficiency". British Journal of Haematology. 141 (3): 298–308. doi:10.1111/j.1365-2141.2008.07017.x. ISSN 1365-2141. PMID 18318771. S2CID 36408296.
- Özsoylu, S. (1972). "Congenital Methemoglobinemia Due to Hemoglobin M". Acta Haematologica. 47 (4): 225–232. doi:10.1159/000208528. ISSN 0001-5792. PMID 4625305.
- Rangan, Aruna; Savedra, Michelle E.; Dergam-Larson, Camila; Swanson, Kenneth C.; Szuberski, Jessica; Go, Ronald S.; Porter, Tavanna R.; Brunker, Sarah E.; Shi, Min; Nguyen, Phuong L.; Hoyer, James D. (August 2021). "Interpreting sulfhemoglobin and methemoglobin in patients with cyanosis: An overview of patients with M-hemoglobin variants". International Journal of Laboratory Hematology. 43 (4): 837–844. doi:10.1111/ijlh.13581. ISSN 1751-553X. PMID 34092029. S2CID 235362094.
- Pisciotta, A. V.; Ebbe, S. N.; Hinz, J. E. (July 1959). "Clinical and laboratory features of two variants of methemoglobin M disease". The Journal of Laboratory and Clinical Medicine. 54 (1): 73–87. ISSN 0022-2143. PMID 13665153.
- Rastogi, Loveena; Langer, Sabina; Radhakrishnan, Nita; Saxena, Renu; Kotwal, Jyoti (2020-09-30). "Hb-M Hyde Park: a rare cause of cyanosis arising from a de novo mutation". Blood Research. 55 (3): 177–180. doi:10.5045/br.2020.2020084. ISSN 2287-979X. PMC 7536569. PMID 32747614.
- Nelson textbook of pediatrics. Robert Kliegman, Bonita Stanton, Joseph W., III St. Geme, Nina Felice Schor, Richard E. Behrman, Waldo E. Preceded by: Nelson (Edition 21 ed.). Philadelphia, PA. 2020. ISBN 978-0-323-56888-3. OCLC 1096283151.
{{cite book}}: CS1 maint: others (link) - Spears, Frances; Banerjee, Arnab (June 2008). "Hemoglobin M variant and congenital methemoglobinemia: methylene blue will not be effective in the presence of hemoglobin M". Canadian Journal of Anaesthesia. 55 (6): 391–392. doi:10.1007/BF03021499. ISSN 0832-610X. PMID 18566207. S2CID 46308991.
- Vichinsky, E. P.; Lubin, B. H. (May 1980). "Unstable hemoglobins, hemoglobins with altered oxygen affinity, and m-hemoglobins". Pediatric Clinics of North America. 27 (2): 421–428. doi:10.1016/s0031-3955(16)33859-7. ISSN 0031-3955. PMID 7383714.
- Nathan and Oski's hematology and oncology of infancy and childhood. Stuart H. Orkin, David E. Fisher, David Ginsburg, A. Thomas Look, Samuel E. Lux, David G. Nathan (8th ed.). Philadelphia, PA. 2015. ISBN 978-0-323-29177-4. OCLC 894113533.
{{cite book}}: CS1 maint: others (link) - Mihailescu, Mihaela-Rita; Russu, Irina M. (2001-03-27). "A signature of the T → R transition in human hemoglobin". Proceedings of the National Academy of Sciences of the United States of America. 98 (7): 3773–3777. Bibcode:2001PNAS...98.3773M. doi:10.1073/pnas.071493598. ISSN 0027-8424. PMC 31128. PMID 11259676.
- Thom, Christopher S.; Dickson, Claire F.; Gell, David A.; Weiss, Mitchell J. (March 2013). "Hemoglobin Variants: Biochemical Properties and Clinical Correlates". Cold Spring Harbor Perspectives in Medicine. 3 (3): a011858. doi:10.1101/cshperspect.a011858. ISSN 2157-1422. PMC 3579210. PMID 23388674.
- Kim, Dae Sung; Baek, Hee Jo; Kim, Bo Ram; Yoon, Bo Ae; Lee, Jun Hyung; Kook, Hoon (December 2020). "The First Korean Family with Hemoglobin-M Milwaukee-2 Leading to Hereditary Methemoglobinemia". Yonsei Medical Journal. 61 (12): 1064–1067. doi:10.3349/ymj.2020.61.12.1064. ISSN 1976-2437. PMC 7700874. PMID 33251782.
- Stucke, Astrid G.; Riess, Matthias L.; Connolly, Lois A. (April 2006). "Hemoglobin M (Milwaukee) affects arterial oxygen saturation and makes pulse oximetry unreliable". Anesthesiology. 104 (4): 887–888. doi:10.1097/00000542-200604000-00036. ISSN 0003-3022. PMID 16571987.