Inclusion body myositis
Inclusion body myositis (IBM) (/maɪoʊˈsaɪtɪs/) (sometimes called sporadic inclusion body myositis, sIBM) is the most common inflammatory muscle disease in older adults.[2] The disease is characterized by slowly progressive weakness and wasting of both proximal muscles (located on or close to the torso) and distal muscles (close to hands or feet), most apparent in the finger flexors and knee extensors.[3] IBM is often confused with an entirely different class of diseases, called hereditary inclusion body myopathies (hIBM).[4][5] The "M" in hIBM is an abbreviation for "myopathy" while the "M" in IBM is for "myositis".[6] In IBM, two processes appear to occur in the muscles in parallel, one autoimmune and the other degenerative. Inflammation is evident from the invasion of muscle fibers by immune cells. Degeneration is characterized by the appearance of holes, deposits of abnormal proteins, and filamentous inclusions in the muscle fibers.[7] sIBM is a rare disease, with a prevalence ranging from 1 to 71 individuals per million.[8][9]
| Inclusion body myositis | |
|---|---|
| Other names | sIBM |
| Specialty | Rheumatology, Neurology, Neuromuscular medicine |
| Symptoms | Weakness |
| Usual onset | Typically after age 45[1] |
| Differential diagnosis | deconditioning, hereditary muscle diseases[1] |
| Frequency | 5-71/100,000,000[1] |
Weakness comes on slowly (over months to years) in an asymmetric manner and progresses steadily, leading to severe weakness and wasting of arm and leg muscles. IBM is more common in men than women.[10] Patients may become unable to perform activities of daily living and most require assistive devices within 5 to 10 years of symptom onset.[11] sIBM does not significantly affect life expectancy,[1] although death related to malnutrition and respiratory failure can occur.[12] The risk of serious injury due to falls is increased.[1] There is no effective treatment for the disease as of 2019.[1]
Classification and terminology
IBM stands for "inclusion body myositis:, not "inclusion body myopathy."[6] The 'inclusion body' refers to a histological finding of rimmed vacules in muscle tissue.[6] However, IBM does not refer to the collection of diseases that feature these inclusion bodies. It refers to a specific disease entity.[6]
Multiple genetic diseases that feature inclusion bodies have been grouped into "hereditary inclusion body myopathies (hIBM)."[6] Myopathy is used because inflammation is not a prominent finding. There is inconsistency in what individual disease entities fall under the category of hIBM.[6] The term "sporadic inclusion body myositis" (sIBM) was introduced as a way to refer to IBM to avoid confusion with hIBM.[6] However, one author discourages use of sIBM, as it implies that IBM and hIBM differ only in inheritance; they actually have unrelated mechanisms and manifestations of disease.[6]
Signs and symptoms
_in_IBM.png.webp)
sIBM causes progressive muscle weakness.[1] How sIBM affects individuals is variable, including the age of onset (which generally varies from the forties upwards) and rate of progression. Because of this variability, there is no "textbook case".[13]
Common early symptoms include frequent tripping and falling and difficulty going up stairs. Foot drop in one or both feet can occur.[14] Part of the cause for this dysfunction is the early involvement of the quadriceps muscles.[1] Weakness of the tibialis anterior muscle is responsible for foot drop. Another common early symptom is trouble manipulating the fingers, such as difficulty with tasks such as turning doorknobs or gripping keys. Weakness of finger flexion and ankle dorsiflexion occurs early.[1] sIBM also preferentially affects the wrist flexors, biceps, and triceps.[1]
During the course of the illness, the patient's mobility is progressively restricted as it becomes difficult to bend down, reach for things, and walk quickly. Many patients say they have balance problems and fall easily, as the muscles cannot compensate for an off-balanced posture. Because sIBM makes the leg muscles weak and unstable, patients are very vulnerable to serious injury from tripping or falling down. Although pain has not been traditionally part of the "textbook" description, many patients report severe muscle pain, especially in the thighs.
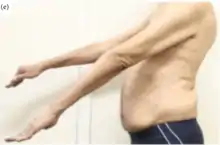
Progressive difficulty swallowing (dysphagia) is present in 40 to 85% of IBM cases and often leads to death from aspiration pneumonia.[15]
IBM can also result in diminished capacity for aerobic exercise. This decline is most likely a consequence of the sedentary lifestyle leading to disuse muscle atrophy that is often associated with the symptoms of IBM (i.e. progressive muscle weakness, decreased mobility, and increased level of fatigue). Therefore, one focus of treatment should be the improvement of aerobic capacity.[16]
Patients with sIBM usually eventually need to resort to a cane or a walker and in most cases, a wheelchair eventually becomes a necessity.
"The progressive course of s-IBM leads slowly to severe disability. Finger functions can become very impaired, such as manipulating pens, keys, buttons, and zippers, pulling handles, and firmly grasping handshakes. Arising from a chair becomes difficult. Walking becomes more precarious. Sudden falls, sometimes resulting in major injury to the skull or other bones, can occur, even from walking on minimally irregular ground or from other minor imbalances outside or in the home, due to weakness of quadriceps and gluteus muscles depriving the patient of automatic posture maintenance. A foot-drop can increase the likelihood of tripping. Dysphagia can occur, usually caused by upper esophageal constriction that often can be symptomatically improved, for several months to years, by bougie dilation per a GI or ENT physician. Respiratory muscle weakness can sometimes eventuate."[17]
Causes
The cause of IBM is unknown. IBM likely results from the interaction of a number of genetic and environmental factors.[18]
There are two major theories about how sIBM is caused. One hypothesis suggests that the inflammation-immune reaction, caused by an unknown trigger – likely an undiscovered virus or an autoimmune disorder – is the primary cause of sIBM and that the degeneration of muscle fibers and protein abnormalities are secondary features.[19] Despite the arguments "in favor of an adaptive immune response in sIBM, a purely autoimmune hypothesis for sIBM is untenable because of the disease's resistance to most immunotherapy."[20]
The second school of thought advocates the theory that sIBM is a degenerative disorder related to aging of the muscle fibers and that abnormal, potentially pathogenic protein accumulations in myofibrils play a key causative role in sIBM (apparently before the immune system comes into play). This hypothesis emphasizes the abnormal intracellular accumulation of many proteins, protein aggregation and misfolding, proteosome inhibition, and endoplasmic reticulum (ER) stress.[17]
One review discusses the "limitations in the beta-amyloid-mediated theory of IBM myofiber injury."[21]
Dalakas (2006) suggested that a chain of events causes IBM – some sort of virus, likely a retrovirus, triggers the cloning of T cells. These T cells appear to be driven by specific antigens to invade muscle fibers. In people with sIBM, the muscle cells display "flags" telling the immune system that they are infected or damaged (the muscles ubiquitously express MHC class I antigens) and this immune process leads to the death of muscle cells. The chronic stimulation of these antigens also causes stress inside the muscle cell in the endoplasmic reticulum (ER) and this ER stress may be enough to cause a self-sustaining T cell response (even after a virus has dissipated). In addition, this ER stress may cause the misfolding of protein. The ER is in charge of processing and folding molecules carrying antigens. In IBM, muscle fibers are overloaded with these major histocompatibility complex (MHC) molecules that carry the antigen protein pieces, leading to more ER stress and more protein misfolding.[19]
A self-sustaining T cell response would make sIBM a type of autoimmune disorder. When studied carefully, it has not been possible to detect an ongoing viral infection in the muscles. One theory is that a chronic viral infection might be the initial triggering factor setting IBM in motion. There have been a handful of IBM cases – approximately 15 – that have shown clear evidence of a virus called HTLV-1. The HTLV-1 virus can cause leukemia, but in most cases lies dormant and most people end up being lifelong carriers of the virus. One review says that the best evidence points towards a connection with some type of retrovirus and that a retroviral infection combined with immune recognition of the retrovirus is enough to trigger the inflammation process.[19]
- amyloid protein
- The hypothesis that beta amyloid protein is key to IBM has been supported in a mouse model using an Aβ vaccine that was found to be effective against inclusion body myositis in mouse models. Although this vaccine is likely, not safe for human use, it still shows that attacking Aβ has efficacy in mice against IBM.[22]
- Following up on earlier leads, the Greenberg group report finding that the protein TDP-43 is a very prominent and highly sensitive and specific feature of IBM. This protein is normally found within the nucleus but in IBM is found in the cytoplasm of the cell. This important advance should help develop a new screening technique for IBM and may provide clues in terms of a therapeutic approach[23]
Genetics
sIBM is not inherited and is not passed on to the children of IBM patients. There are genetic features that do not directly cause IBM but that appear to predispose a person to getting IBM – having this particular combination of genes increases one's susceptibility to getting IBM. Some 67% of IBM patients have a particular combination of human leukocyte antigen genes in a section of the 8.1 ancestral haplotype in the center of the MHC class II region. sIBM is not passed on from generation to generation, although the susceptibility region of genes may be.[19]
There are also several rare forms of hereditary inclusion body myopathy that are linked to specific genetic defects and that are passed on from generation to generation. Since these forms do not show features of muscle inflammation, they are classified as myopathies rather than forms of myositis. Because they do not display inflammation as a primary symptom, they may in fact be similar, but different diseases to sporadic inclusion body myositis. There are several different types, each inherited in different ways. See hereditary inclusion body myopathy.
A 2007 review concluded there is no indication that the genes responsible for the familial or hereditary conditions are involved in sIBM.[24]
Diagnosis
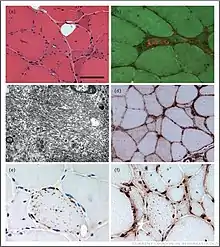
Elevated creatine kinase (CK) levels in the blood (at most ~10 times normal) are typical in sIBM but affected individuals can also present with normal CK levels. Electromyography (EMG) studies display variable abnormalities such as increased insertional activity[25], increased spontaneous activity (fibrillation potentials and sharp waves),[1] and large/broad or short/narrow motor unit potentials.[1] On EMG, recruitment patterns can be reduced or increased. Findings can vary even within the same muscle of an affected individual.[1] Muscle biopsy may display several common findings including; inflammatory cells invading muscle cells, vacuolar degeneration, and inclusion bodies of aggregations of multiple proteins.[26] sIBM is a challenge to the pathologist and even with a biopsy, diagnosis can be ambiguous.[27]
A diagnosis of inclusion body myositis was historically dependent on muscle biopsy results. Antibodies to cytoplasmic 5'-nucleotidase (cN1A; NT5C1A) have been strongly associated with the condition.[1] However, other inflammatory conditions, such as lupus, can have a positive anti-NT5C1A.[1] As of 2019, it remains to be established whether a positive anti-NT5C1A antibody test can make muscle biopsy unneeded.[1]
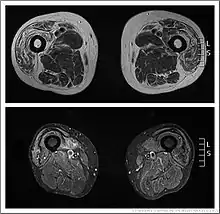
Muscle imaging can help establish the pattern of muscle involvement and selection of a biopsy site.[1]
Differential diagnosis
IBM is often initially misdiagnosed as polymyositis. A course of prednisone is typically completed with no improvement and eventually, sIBM is confirmed. sIBM weakness comes on over months or years and progresses steadily, whereas polymyositis has an onset of weeks or months. Muscular dystrophy (e.g. limb girdle muscular dystrophy) must be considered as well. sIBM can be mistaken for physical deconditioning.[1]
Hereditary myopathies can mimic sIBM, both in signs and symptoms and in the appearance of muscle biopsies. A small percentage of those initially diagnosed with sIBM are later found to have pathogenic mutations in the genes VCP and SQSTM1, which are known to cause hIBM.[1]
IBM has a distinctive pattern of muscle involvement that distinguishes it among inflammatory myopathies.[6] Characteristic of IBM is weakness of finger flexion, knee extension, and ankle dorsiflexion.[6] Other inflammatory myopathies cause a proximal muscle weakness pattern, such as weakness of hip flexion, abduction, and extension, as well as shoulder abduction.[6] IBM and other inflammatory myopathies both cause bicep/tricep weakness.[6]
Classification
- The common type is sIBM; it strikes individuals apparently at random.[28]
- There is a type that has been observed in multiple siblings in the same generation in several families, termed familial inflammatory sIBM, but it is not passed on from generation to generation.[19]
- There are also several very rare forms of hereditary inclusion body myopathy (hIBM) that are linked to specific genetic defects and that are passed on from generation to generation, each inherited in different ways.[29]
Management
There is no standard course of treatment to slow or stop the progression of the disease as of 2019.[1] sIBM patients do not reliably respond to anti-inflammatory, immunosuppressant, or immunomodulatory medications.[30] Most of disease management is supportive care.[1] Prevention of falls is an important consideration.[1] There is no consensus on exercise guidelines; however, physical therapy is recommended to teach the patient a home exercise program, to teach how to compensate during mobility-gait training with an assistive device, transfers and bed mobility.[31] An exercise regimen preferentially minimizes a patient's risk of injury and corresponds to the patient's goals.[1]
Other related disorders
When sIBM was originally described, the major feature noted was muscle inflammation. Two other disorders were also known to display muscle inflammation, and sIBM was classified along with them. They are dermatomyositis (DM) and polymyositis (PM) and all three illnesses were called idiopathic (of unknown origin) myositis or inflammatory myopathies.[32]
It appears that sIBM and polymyositis share some features, especially the initial sequence of immune system activation, however, polymyositis comes on over weeks or months, does not display the subsequent muscle degeneration and protein abnormalities as seen in IBM, and as well, polymyositis tends to respond well to treatments, IBM does not. IBM is often confused with (misdiagnosed as) polymyositis. Polymyositis that does not respond to treatment is likely IBM.[33]
Dermatomyositis shares a number of similar physical symptoms and histopathological traits as polymyositis, but exhibits a skin rash not seen in polymyositis or sIBM. It may have different root causes unrelated to either polymyositis or sIBM.[34]
Mutations in valosin-containing protein (VCP) cause multisystem proteinopathy (MSP), which can present (among others) as a rare form of inclusion body myopathy.[35]
Epidemiology
Prevalence of disease in a rigorous meta-analysis in 2017 was 46 patients per million.[6] The earliest published prevalence was in 2000 and put at 5 per million.[6] A 2017 study in Ireland reported 112 per million.[6] It is not believed that the disease prevalence is increasing with time, but rather diagnostics and reporting are improving.[6]
Estimates of the mean age of onset range from 61 to 68 years old.[6]
Society and culture
In the biographical drama film Father Stu, the protagonist, a boxer-turned-Catholic priest, has sIBM.[36][37]
See also
References
- Weihl, CC (December 2019). "Sporadic Inclusion Body Myositis and Other Rimmed Vacuolar Myopathies". Continuum (Minneapolis, Minn.). 25 (6): 1586–1598. doi:10.1212/CON.0000000000000790. PMID 31794461. S2CID 208531761.
- Ahmed, Mhoriam; Machado, Pedro M; Miller, Adrian; Spicer, Charlotte; Herbelin, Laura; et, al (March 23, 2016). "Targeting protein homeostasis in sporadic inclusion body myositis". Science Translational Medicine. 8 (331): 331ra41. doi:10.1126/scitranslmed.aad4583. PMC 5043094. PMID 27009270.
- Jackson, CE; Barohn, RJ; Gronseth, G; Pandya, S; Herbelin, L; and, The Muscle Study Group (April 2008). "Inclusion body myositis functional rating scale: a reliable and valid measure of disease severity". Muscle and Nerve. 37 (4): 473–476. doi:10.1002/mus.20958. PMID 18236463. S2CID 22284747.
- IBMmyositis.com
- cureibm.org
- Greenberg, SA (May 2019). "Inclusion body myositis: clinical features and pathogenesis". Nature Reviews. Rheumatology. 15 (5): 257–272. doi:10.1038/s41584-019-0186-x. PMID 30837708. S2CID 71146208.
- Machado, P; Dimachkie, MM; Bahron, RJ (October 2014). "Sporadic Inclusion Body Myositis: new insights and potential therapy". Current Opinion in Neurology. 27 (5): 591–598. doi:10.1097/WCO.0000000000000129. PMC 4248565. PMID 25159931.
- Machado, P; Brady, S; Hanna, MG (2013). "Update in inclusion body myosities". Current Opinion in Rheumatology. 25 (763–771): 763–771. doi:10.1097/01.bor.0000434671.77891.9a. PMC 4196838. PMID 24067381.
- "Sporadic Inclusion Body Myositis".
- "Inclusion Body Myositis. IBM information; Age Related illness".
- "Understanding IBM".
- Cox, FM; Titulaer, MJ; Sont, JK; Wintzen, AR; et, al (November 1, 2011). "A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities". Brain. 134 (11): 3167–3175. doi:10.1093/brain/awr217. PMID 21908393.
- Evangelista, Teresinha; Weihl, Conrad C.; Kimonis, Virginia; Lochmüller, Hanns; Clemen, Christoph; Deshaies, Ray; Evangelista, Teresinha; Eymard, Bruno; Greensmith, Linda; Hilton-Jones, David; Kimonis, Virginia; Kley, Rudy; Lochmüller, Hanns; Meyer, Hemmo; Mozaffar, Tahseen (August 2016). "215th ENMC International Workshop VCP-related multi-system proteinopathy (IBMPFD) 13–15 November 2015, Heemskerk, The Netherlands". Neuromuscular Disorders. 26 (8): 535–547. doi:10.1016/j.nmd.2016.05.017. ISSN 0960-8966.
- "Inclusion Body Myositis". www.google.com. Retrieved 2021-06-12.
{{cite web}}: CS1 maint: url-status (link) - Oh TH, Brumfield KA, Hoskin TL, Kasperbauer JL, Basford JR (2008). "Dysphagia in inclusion body myositis: clinical features, management, and clinical outcome". Am J Phys Med Rehabil. 87 (11): 883–9. doi:10.1097/PHM.0b013e31818a50e2. PMID 18936555. S2CID 11878442.
- Johnson LG, Collier KE, Edwards DJ, et al. (June 2009). "Improvement in aerobic capacity after an exercise program in sporadic inclusion body myositis". J Clin Neuromuscul Dis. 10 (4): 178–84. doi:10.1097/CND.0b013e3181a23c86. PMID 19494728. S2CID 14686189.
- Askanas V, Engel WK (2006). "Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition". Neurology. 66 (2 Suppl 1): S39–S48. doi:10.1212/01.wnl.0000192128.13875.1e. PMID 16432144. S2CID 24365234.
- "Inclusion Body Myositis (IBM)". Retrieved 7 May 2017.
- Dalakas MC (2006). "Sporadic inclusion body myositis--diagnosis, pathogenesis and therapeutic strategies". Nat Clin Pract Neurol. 2 (8): 437–447. doi:10.1038/ncpneuro0261. PMID 16932602. S2CID 7462545.
- Inclusion Body Myositis at eMedicine
- Greenberg SA. (2009). "Inclusion body myositis: review of recent literature". Curr Neurol Neurosci Rep. 9 (1): 83–89. doi:10.1007/s11910-009-0013-x. PMID 19080758. S2CID 16391053.
- Kitazawa M, Vasilevko V, Cribbs DH, LaFerla FM (13 May 2009). "Immunization with amyloid-β attenuates inclusion body myositis-like myopathology and motor impairment in a transgenic mouse model". The Journal of Neuroscience. 29 (19): 6132–41. doi:10.1523/JNEUROSCI.1150-09.2009. PMC 3049190. PMID 19439591.
Inclusion body myositis...features include T-cell mediated inflammatory infiltrates and aberrant accumulations of proteins, including amyloid-β (Aβ), tau, ubiquitinated proteins, apolipoprotein E, and β-synuclein in skeletal muscle. ... active immunization markedly reduces intracellular Aβ deposits and attenuates the motor impairment compared with untreated mice...Aβ oligomers contribute to the myopathy process as they were significantly reduced in the affected skeletal muscle from immunized mice. In addition, the anti-Aβ antibodies produced in the immunized mice blocked the toxicity of the Aβ oligomers in vitro, providing a possible key mechanism for functional recovery.
- Salajegheh, M, Pinkus, JL, Taylor, JP, Amato, AA, Nazareno, R, Baloh, RH, Greenberg, SA. (2009). "Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis". Muscle Nerve. 40 (1): 19–31. doi:10.1002/mus.21386. PMC 2700211. PMID 19533646.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Needham M, Mastaglia FL, Garlepp MJ (2007). "Genetics of inclusion-body myositis". Muscle Nerve. 35 (5): 549–561. doi:10.1002/mus.20766. PMID 17366591. S2CID 23665612.
- Moghadam-Kia, Siamak; Oddis, Chester V.; Aggarwal, Rohit (2016). "Approach to asymptomatic creatine kinase elevation". Cleveland Clinic Journal of Medicine. 83 (1): 37–42. doi:10.3949/ccjm.83a.14120. ISSN 0891-1150.
- "Sporadic Inclusion Body Myositis". NORD (National Organization for Rare Disorders).
- "muscle biopsy revealed: Topics by Science.gov". www.science.gov. Retrieved 2022-10-28.
- Karpati G, O'Ferrall EK (Jan 2009). "Sporadic inclusion body myositis: Pathogenic considerations". Ann Neurol. 65 (1): 7–11. doi:10.1002/ana.21622. PMID 19194875. S2CID 41203262.
- Broccolini A.; Mirabella M. (2014). "Hereditary inclusion-body myopathies". Biochim. Biophys. Acta. 1852 (4): 644–650. doi:10.1016/j.bbadis.2014.08.007. PMID 25149037.
- Suzuki, Naoki; Mori-Yoshimura, Madoka; Yamashita, Satoshi; Nakano, Satoshi; Murata, Ken-ya; Inamori, Yukie; Matsui, Naoko; Kimura, En; Kusaka, Hirofumi; Kondo, Tomoyoshi; Higuchi, Itsuro; Kaji, Ryuji; Tateyama, Maki; Izumi, Rumiko; Ono, Hiroya (2016-11-08). "Multicenter questionnaire survey for sporadic inclusion body myositis in Japan". Orphanet Journal of Rare Diseases. 11 (1). doi:10.1186/s13023-016-0524-x. ISSN 1750-1172.
- "Inclusion Body Myositis Information Page". National Institute of Neurological Disorders and Stroke. March 17, 2019. Retrieved June 25, 2021.
{{cite web}}: CS1 maint: url-status (link) - "Dermatomyositis (DM)". The Lecturio Medical Concept Library. Retrieved 11 July 2021.
- When myositis doesn't respond to treatment Retrieved 20 April 2015.
- Malik, Asma; Hayat, Ghazala; Kalia, Junaid S.; Guzman, Miguel A. (2016-05-20). "Idiopathic Inflammatory Myopathies: Clinical Approach and Management". Frontiers in Neurology. 7. doi:10.3389/fneur.2016.00064. ISSN 1664-2295.
- Harrison AF, Shorter J (April 2017). "RNA-binding proteins with prion-like domains in health and disease". Biochem. J. 474 (8): 1417–1438. doi:10.1042/BCJ20160499. PMC 5639257. PMID 28389532.
- Fleming, Kristen (13 April 2022). "'Father Stu': How a motorcycle accident turned this former boxer into a priest". New York Post. Archived from the original on 22 April 2022. Retrieved 27 April 2022.
- Rapold, Nicolas (13 April 2022). "'Father Stu' Review: Screwball Salvation". The New York Times. Archived from the original on 16 April 2022. Retrieved 27 April 2022.