JTV-519
JTV-519 (K201) is a 1,4-benzothiazepine derivative that interacts with many cellular targets.[1] It has many structural similarities to diltiazem, a Ca2+ channel blocker used for treatment of hypertension, angina pectoris and some types of arrhythmias.[2] JTV-519 acts in the sarcoplasmic reticulum (SR) of cardiac myocytes by binding to and stabilizing the ryanodine receptor (RyR2) in its closed state.[3][4] It can be used in the treatment of cardiac arrhythmias, heart failure, catecholaminergic polymorphic ventricular tachycardia (CPVT) and store overload-induced Ca2+ release (SOICR).[2][3][4] Currently, this drug has only been tested on animals and its side effects are still unknown.[5] As research continues, some studies have also found a dose-dependent response; where there is no improvement seen in failing hearts at 0.3 μM and a decline in response at 1 μM.[4]
 | |
| Names | |
|---|---|
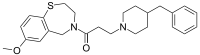
| Preferred IUPAC name
3-(4-Benzylpiperidin-1-yl)-1-(7-methoxy-2,3-dihydro-1,4-benzothiazepin-4(5H)-yl)propan-1-one | |
| Other names
K201 | |
| Identifiers | |
CAS Number |
|
3D model (JSmol) |
|
| ChemSpider |
|
| ECHA InfoCard | 100.236.521 |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
SMILES
| |
| Properties | |
Chemical formula |
C25H32N2O2S |
| Molar mass | 424.60 g·mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |
Treatment with JTV-519 involves stabilization of RyR2 in its closed state, decreasing its open probability during diastole and inhibiting a Ca2+ leak into the cell's cytosol.[3][4] By decreasing the intracellular Ca2+ leak, it is able to prevent Ca2+ sparks or increases in the resting membrane potential, which can lead to spontaneous depolarization (cardiac arrhythmias), and eventually heart failure, due to the unsynchronized contraction of the atrial and ventricular compartments of the heart.[2][3][4] When Ca2+ sparks occur from the SR, the increase in intracellular Ca2+ contributes to the rising membrane potential which leads to the irregular heart beat associated to cardiac arrhythmias.[3] It can also prevent SOICR in the same manner; preventing opening of the channel due to the increase of Ca2+ inside the SR levels beyond its threshold.[2]
Molecular problem
In the closed state, N-terminal and central domains come into close contact interacting to cause a “zipping” of domains. This leads to conformational constraints that stabilize the channel and maintain the closed state.[1] Most RyR2 mutations are clustered into three regions of the channel, all affecting the same domains that interact to stabilize the channel.[1] Any of these mutations can lead to “unzipping” of the domains and a decrease in the energy barrier required for opening the channel (increasing its open probability).[1] This channel “unzipping” allows for an increase in protein kinase A phosphorylation and calstabin2 dissociation. Phosphorylation of RyR2 increases the channel's response to Ca2+, which usually binds the RyR2 to open it.[1] If the channel become phosphorylated, this can lead to an increase in Ca2+ sparks due to an increase in Ca2+ sensitivity.
Some researchers believe that the depletion of calstabin2 from the RyR2 causes the calcium leak.[3] The depletion of calstabin2 can occur in both heart failure and CPVT.[3] Calstabin2 is a protein that stabilizes RyR2 in its closed state, preventing Ca2+ leakage during diastole. When calstabin2 is lost, the interdomain interactions of RyR2 become loose, allowing the Ca2+ leak.[3] However, the role of calstabin2 has been controversial, as some studies have found it necessary for the effect of JTV-519,[3] whereas others have found the drug functions without the stabilizing protein.[2]
Molecular mechanism
JTV-519 seems to restore the stable conformation of RyR2 during the closed state.[1][4] It is still controversial whether or not calstabin2 is necessary for this process, however, many studies believe that JTV-519 can act directly on the channel and by binding, prevents conformational changes.[2] This stabilization of the channel decreases its open probability resulting in fewer leaks of Ca2+ into the cytosol and fewer Ca2+ sparks to occur.[3][4] Researchers who believe that calstabin2 is necessary for JTV-519 effect, found that this drug may function by inducing the binding of calstabin2 back to the channel or increasing calstabin2's affinity for the RyR2 and thus increasing its stability.[2][3]
References
- Oda, T; Yano, M; Yamamoto, T; Tokuhisa, T; Okuda, S; Doi, M; Ohkusa, T; Ikeda, Y; et al. (2005). "Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure". Circulation. 111 (25): 3400–10. doi:10.1161/CIRCULATIONAHA.104.507921. PMID 15967847.
- Hunt, DJ; Jones, PP; Wang, R; Chen, W; Bolstad, J; Chen, K; Shimoni, Y; Chen, SR (2007). "K201 (JTV519) suppresses spontaneous Ca2+ release and 3Hryanodine binding to RyR2 irrespective of FKBP12.6 association". The Biochemical Journal. 404 (3): 431–8. doi:10.1042/BJ20070135. PMC 1896290. PMID 17313373.
- Wehrens, XH; Lehnart, SE; Reiken, SR; Deng, SX; Vest, JA; Cervantes, D; Coromilas, J; Landry, DW; Marks, AR (2004). "Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2". Science. 304 (5668): 292–6. doi:10.1126/science.1094301. PMID 15073377. S2CID 41980576.
- Toischer, K; Lehnart, SE; Tenderich, G; Milting, H; Körfer, R; Schmitto, JD; Schöndube, FA; Kaneko, N; et al. (2010). "K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum". Basic Research in Cardiology. 105 (2): 279–87. doi:10.1007/s00395-009-0057-8. PMC 2807967. PMID 19718543.
- Viswanathan, MN; Page, RL (2009). "Pharmacological therapy for atrial fibrillation: Current options and new agents". Expert Opinion on Investigational Drugs. 18 (4): 417–31. doi:10.1517/13543780902773410. PMID 19278302. S2CID 71155277.