Lateral meningocele syndrome
Lateral meningocele syndrome, also known as Lehman syndrome,[2] is a very rare skeletal disorder with facial anomalies, hypotonia, and meningocele-related neurologic dysfunction.[3] These protrusions form from membranes surrounding the spinal cord in gaps in the spine (vertebrae).[4] They most often occur in the lower spine and damage the surrounding nerves that spread throughout the rest of the body.[4] Examples of resulting damages are bladder function, prickling or tingling sensations, stiffness and weakness in the legs, and back pain.[4] People affected with lateral meningocele typically have high arched eyebrows, widely spaced eyes, droopy eyes, and other facial features. There have been only 14 reported individuals with lateral meningocele syndrome with 7 of those who have a molecularly confirmed diagnosis.[5] There is no specific treatment for this syndrome, but only supportive management including lateral spinal meningoceles, psychomotor development, musculoskeletal, and routine management.[5]
| Lateral meningocele syndrome | |
|---|---|
| Other names | Lehman syndrome[1] |
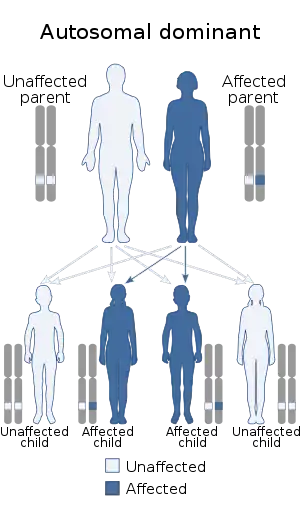 | |
| Lateral meningocele syndrome is inherited in an autosomal dominant manner | |
Signs and Symptoms
Facial features found in this syndrome include[6]
- dolichocephaly
- hypertelorism
- ptosis
- microretrognathia
- high arched palate
- long flat philtrum
- low set ears
Non facial features of this syndrome include[6]
- hyperextensibility
- hypotonia
- lateral meningoceles
- bladder dysfunction and neuropathy
This syndrome also leads to a delayed development of motor skills in infancy, including sitting and crawling. Intelligence, however, usually stays unaffected.[6] Some other features of this syndrome are low muscle tone during infancy, decreased muscle bulk, loose joints, and hernias.[6]
Diagnosis is based on a presentation concurrent with previous clinical reports, as well as a heterozygous pathogenic variant in the NOTCH3 gene.[7]
Diagnosis
Diagnosis of Lehman syndrome may be suspected based on several distinctive facial features, the presence of lateral meningoceles, hyperextensibility, and hypotonia.[8] Aside from physical presence, radiographic images of the spine may also clinically diagnose lateral meningoceles. With molecular genetic testing, Lehman syndrome is positively identified with the presence of a pathogenic variant in NOTCH3.[9] When the disorder was initially discovered, features of maldevelopment of the spinal cord, cerebellum, and cerebral cortex distinguished the diagnosis of Lehman syndrome.[10] Among all historical cases (from the period 1997-2015), patients were diagnosed with disorder before the age of 20 years old and as early as 5 years old.[11][8]
Treatment
Currently, there is no treatment for Lehman syndrome. The only suggestions for patients with this disorder is to manage any of the associated symptoms.[9] For instance, pain management options are present for those experiencing chronic pain. Under rare circumstances, surgical intervention is required for neurologic manifestations. Further, some patient benefit from rehabilitation medicine, physiotherapy, as well as routine management of cleft palate, hearing loss, congenital cardiac defects, genitourinary abnormalities, and feeding difficulties.[7]
History
This syndrome was first described by Lehman et al. in 1977.[12] This paper described a 14-year-old girl with a number of unusual findings. Her mother shared some of the same findings. Since then over a dozen additional cases have been reported.
Genetics
This syndrome appears to be inherited in an autosomal dominant fashion; however, X-linked inheritance has not been completely ruled out.[13] Males diagnosed with Lehman syndrome were effected the same as females causing the believed inheritance pattern to be autosomal dominant, not X-linked.[6]
Lehman syndrome is associated with the heterozygous truncating mutation on exon33 of the Notch 3 gene located on chromosome 19p13.[6] The mutation was found using whole-exome sequencing and confirmed with Sanger sequencing.
Molecular analyses suggest that the causative mutations cause a truncation of the protein. These mutations result in the loss of PEST sequence in the protein. This loss is associated with a prolonged half life of the protein and therefore an increase in signaling effects.[6]
Mutations in Notch 3 were found to be associated with this syndrome.[6] Notch 3 produces the Notch 3 protein,[14] a receptor protein in which ligands bind to control gene activity in a cell's nucleus. The Notch 3 gene is a part of the Notch family of genes which are associated with cell differentiation and function.[15] The Notch 3 mutation is a truncating mutation because it results in the loss of the intracellular end of the Notch 3 protein known as the Notch 3 intracellular domain, NICD.[14] This section of the truncated NCID is responsible for disintegration of the NCID after it has gone into the nucleus and completed its function.[4] The mutation causes the Notch 3 protein to be in the cell nucleus for a prolonged period of time continuing to affect gene activity.[14]
Epidemiology
This genetic disease in very rare. There have been 16 reported cases in 14 different families.[4] 9 cases were male patients and 7 cases were female patients.[4]
Inheritance of Lehman syndrome from a parent has occurred. However de novo mutations of the Notch 3 gene are more common.[4]
There is no prevalence in specific populations.[5]
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Lateral meningocele syndrome". www.orpha.net. Retrieved 20 October 2019.
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Lehman syndrome". www.orpha.net. Retrieved 2021-04-21.
- Pamir, M. Memet Özek, Giuseppe Cinalli, Wirginia J. Maixner; forewords by C. Sainte-Rose, C. di Rocco; preface by M. Necmettin, ed. (2008). Spina bifida : management and outcome. Milan: Springer. p. 432. ISBN 9788847006508.
- "Lateral meningocele syndrome: MedlinePlus Genetics". medlineplus.gov. Retrieved 2022-03-17.
- Ejaz, Resham; Carter, Melissa; Gripp, Karen (1993). "NOTCH3-Related Lateral Meningocele Syndrome". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.). Lateral Meningocele Syndrome. GeneReviews®. Seattle (WA): University of Washington, Seattle. PMID 27336130. Retrieved 2022-03-17.
- "OMIM Entry - # 130720 - LATERAL MENINGOCELE SYNDROME; LMNS". www.omim.org. Retrieved 2022-03-17.
- Ejaz, Resham; Carter, Melissa; Gripp, Karen (1993). "NOTCH3-Related Lateral Meningocele Syndrome". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.). Lateral Meningocele Syndrome. GeneReviews®. Seattle (WA): University of Washington, Seattle. PMID 27336130. Retrieved 2021-04-21.
- Gripp, Karen W.; Robbins, Katherine M.; Sobreira, Nara L.; Witmer, P. Dane; Bird, Lynne M.; Avela, Kristiina; Makitie, Outi; Alves, Daniela; Hogue, Jacob S.; Zackai, Elaine H.; Doheny, Kimberly F. (February 2015). "Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome". American Journal of Medical Genetics. Part A. 167A (2): 271–281. doi:10.1002/ajmg.a.36863. ISSN 1552-4833. PMC 5589071. PMID 25394726.
- Ejaz, Resham; Carter, Melissa; Gripp, Karen (1993). "NOTCH3-Related Lateral Meningocele Syndrome". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.). Lateral Meningocele Syndrome. GeneReviews®. Seattle (WA): University of Washington, Seattle. PMID 27336130. Retrieved 2022-03-24.
- Lehman, R. A.; Stears, J. C.; Wesenberg, R. L.; Nusbaum, E. D. (January 1977). "Familial osteosclerosis with abnormalities of the nervous system and meninges". The Journal of Pediatrics. 90 (1): 49–54. doi:10.1016/s0022-3476(77)80763-4. ISSN 0022-3476. PMID 830893.
- Philip, N.; Andrac, L.; Moncla, A.; Sigaudy, S.; Zanon, N.; Lena, G.; Choux, M. (October 1995). "Multiple lateral meningoceles, distinctive facies and skeletal anomalies: a new case of Lehman syndrome". Clinical Dysmorphology. 4 (4): 347–351. doi:10.1097/00019605-199510000-00011. ISSN 0962-8827. PMID 8574426. S2CID 1687286.
- Lehman RAW, Stears JC, Wesenberg RL, Nusbaum ED (1977) Familial osteosclerosis with abnormalities of the nervous system and meninges. J Pediat 90: 49-54
- "OMIM Entry - # 130720 - LATERAL MENINGOCELE SYNDROME; LMNS". www.omim.org. Retrieved 2022-03-18.
- "Lateral meningocele syndrome: MedlinePlus Genetics". medlineplus.gov. Retrieved 2022-03-18.
- Canalis, Ernesto (2020). "The Skeleton of Lateral Meningocele Syndrome". Frontiers in Genetics. 11: 620334. doi:10.3389/fgene.2020.620334. ISSN 1664-8021. PMC 7841456. PMID 33519922.