Low-grade fibromyxoid sarcoma
Low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade sarcoma first described by H. L. Evans in 1987.[1] LGFMS are soft tissue tumors of the mesenchyme-derived connective tissues; on microscopic examination, they are found to be composed of spindle-shaped cells that resemble fibroblasts.[2] These fibroblastic, spindle-shaped cells are neoplastic cells that in most cases of LGFMS express fusion genes, i.e. genes composed of parts of two different genes that form as a result of mutations.[2] The World Health Organization (2020) classified LGFMS as a specific type of tumor in the category of malignant fibroblastic and myofibroblastic tumors.[3]
| Low-grade fibromyxoid sarcoma | |
|---|---|
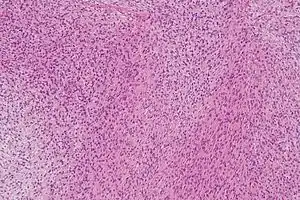 | |
| Micrograph of a low-grade fibromyxoid sarcoma. H&E stain. | |
| Specialty | Pathology |
LGFMS tumors occur in individuals of almost any age but up to 20% are less than 18 years/old. The tumors typically involve the proximal extremities but can occur virtually anywhere in the body.[4] The progression of these tumors commonly takes an indolent, prolonged course involving many years or decades. Over this time, however, the tumors often recur at the site of their surgical removal and/or metastasize usually to the lung or pleural tissues surrounding the lungs.[5] These metastasis can develop decades after the tumor's initial presentation and diagnosis.[6]
LGFMS can be difficult to distinguish from other mesenchymal tumors,[7] particularly from sclerosing epithelioid fibrosarcoma (SEF).[8] LGFMS tumors present with many clinical and pathological features that are similar to those in SEF. Indeed, current studies suggest that LGFMS may be an early form of SEF. For example, a tumor may present with features typical of LGFMS but over time progress to features typical of sclerosing epithelioid fibrosarcomas. This progression may be particularly evident in recurrent or metastatic "LGFMS" tumors.[8][9] Since the World Health Organization has classified LGFMS as one of the malignant fibroblastic and myofibroblastic tumors that is distinctly different than SEF,[3] SEF and LGFMS are here regarded as separate tumor forms.
Presentation
Individuals initially diagnosed with LGFMS had been characterized as young adults (mean age: 33 years) or children >5 years old (13% to 19% of all cases),[10] although a more recent study reported that individuals first diagnosed with the disease ranged in age between 6 and 67 years (mean age: 45.6 year).[8] LGFMS generally presents as a painless mass located in the subcutaneous or subfacial (i.e. beneath the skin) tissues of the upper or lower limbs, trunk,[4] or, less commonly, the head and neck areas or within the gastrointestinal tract, heart, kidney, brain,[11] retroperitoneum, mediastinum, or abdominal cavity.[4] Tumors occurring in subcutaneous and subfacial tissues are often 3–11 cm in size (largest diameter)[8] but in sites where the tumor can go undetected for long periods such as the buttocks,[8] mediastinum,[12] or abdominal cavity[13] have been reported to be as large as 25, 23.5, and 50 cm, respectively. Magnetic resonance imaging and computed tomography scans often give results suggesting that a tumor is a LGFMS.[4][14]
Pathology
LGMFS tumors typically contain one or multiple nodules embedded in a grey-white whorled cut surface. They appear to be infiltrating adjacent structures/tissues in a minority of cases. Microscopic histopathologic views of hematoxylin and eosin stained tumor tissue show whorled and bundled, uniform, bland-appearing, slender fibroblastic spindle-shaped cells with elongated, tapered nuclei. The cells have scant cytoplasm and do not appear to be rapidly proliferating as defined by their low rate of mitosis. The spindle-shaped cells typically occur in an alternating fibrous and myxoid (i.e. more blue or purple compared to normal connective tissue because of excessive uptake of the hematoxylin stain) background. However, the tissues may contain sites dominated by epithelioid cells and other elements that resemble,[2] or are indistinguishable from,[5] sclerosing epithelioid fibrosarcoma. This hybrid pattern is more often seen in recurrent and metastatic LGGMS tumors, may progress to dominate the tissues, and may therefore warrant changing the diagnosis of LGFMD to sclerosing epithelioid fibrosarcoma.[2] While not definitive, LGMFS tumors that are dominated with cells that have an undifferentiated or immature round-cell morphology may be more aggressive than tumors with the typical cell morphology.[2]
Immunohistochemical analyses of LGFMS tumors shows cells that express: MUC1 (also termed EMA), CD99, and/or bcl-2 proteins in some cases; CD34 and SMA proteins in rare cases; and S100, desmin, caldesmon, cytokeratin, and CD117 proteins in virtually no cases.[10] However, MUC4 protein is express by the tumor cells of almost all LGFMS cases[10] and therefore is commonly used as a sensitive and specific marker for LGFMS.[4][5][10] (Since a small subset of LGFMS tumors have been reported to consist of cells that are MUC4-negative, the diagnosis of LGFMS should not be absolutely dependent on finding MUC4-positive tumor cells.[8])
Gene abnormalities
The neoplastic cells in LGFMS tumors express one of various fusion genes. Fusion genes are abnormal genes consisting of parts of two different genes that merge together as a result of large scale gene mutations such as chromosomal translocations, interstitial deletions, or inversions. The fusion genes in LGFMS merge part of the FUS or EWSR2 gene (which is one of the FET protein family genes) with part of a CREB3L2 (i.e. cAMP responsive element binding protein 3 like 2 gene[15]) or CREB3L1 (i.e. CAMP responsive element binding protein 3 like 1 gene). A FUS-CREB3L2 fusion gene is expressed in the majority of LGMFS cases while a FUS-CREB3L1,[16] EWSR1-CREB3L2, or EWSR2-CREBL1[2] fusion gene is expressed in a minority of LGMFS cases. Fusion genes containing part of an AFET gene family gene occur in a wide range of tumors and are though to contribute to the development of at least some of these tumors.[17][18] While the role of the cited fusion genes in the development of LGFMS is not known, the presence of one of these fusion genes is helpful in diagnosing the disease.[10][13]
Diagnosis
The diagnosis of LGFMS rest primary on its tumors' histology consisting of spindle-shaped fibroblastic cells in the appropriate background that express the MUC4 protein and either a FUS-CREB3L2, FUS-CREB3L1, EWSR1-CREB3L2, or EWSR2-CREBL1 fusion gene in most cases.[8] Magnetic resonance imaging and computed tomography scan findings ma also help in the diagnosis.[4] These findings can differentiate LGFMS from various spindle-shaped cell and myxoid neoplasms including benign soft tissue tumors, malignant soft tissue tumors (e.g. desmoid fibromatosis), and sarcomas (eg, malignant peripheral nerve sheath tumora, low-grade myxofibrosarcoma, myxoid dermatofibrosarcoma protuberans, or in rare cases, extraskeletal myxoid chondrosarcoma, synovial sarcomas that have a prominent myxoid background, or myxoid liposarcoma.[8] However, sclerosing epithelioid fibrosarcoma tumors may contain areas that are histopathologically indistinguishable from LGFMS[2] and contain neoplastic cells that expression MUC4 protein in most cases,[19] and a EWSR1-CREB3L1 fusion gene in >20% of cases, a FUS-CREB3L2 fusion gene in ~10% of cases, or a EWSR1-CREB3L2 fusion gene in rare cases.[20] Based on the relative rates of expression in the two tumor types, the presence of tumor neoplastic cells that: 1) express the MUC4 marker protein and a FUS-CREB3L2 fusion gene or an otherwise rearranged FUS gene strongly suggest that the tumor is a SEF;[21] 2) express the EWSR1-CREB3L1 fusion gene suggest that the tumor is a SEF [20] but in any case is unlikely to be a low-grade fibromyxoid sarcoma.[21] 3) express a FUS-CREB3L2 fusion gene is more likely to be a low-grade fibromyxoid sarcoma than a SEF (i.e. the FUS-CREB3L2 fusion gene is present in >90% of low-grade fibromyxoid sarcomas but only ~20% of SEF tumor cells};[22] or 4) express an EWSR1-CREB3L1 or EWSR1-CREB3L2 fusion gene is likely to be a SEF tumor.[19]
Treatment and prognosis
The generally recommended treatment for LGFMS is aggressive, wide surgical excision of the tumor[8] including even those located in difficult to access sites such as the mediastinum.[12] Complete removal of all tumor tissue is critical in lowering the rates of recurrences and metastases.[6] Over the long term, surgically resected LGFMS tumors have tended to recur at the site or resection in up to 75% of cases.[11] These recurrences can develop as long as 15 years (median: 3.5 years) after the initial diagnosis of the disease.[6] Following initial surgical resection, metastases have been reported to develop in up to 50% of individuals.[11] These metastases have been diagnosed up to 45 years (median: 5 years) after the initial diagnosis of LGFMS. They were found mostly in the lungs, pleura, and chest wall, occasionally in the pericardium (i.e. the sac containing the heart), and rarely in a bone, the liver, or the heart.[6] Recurrences of the tumors at the site of their resection as well as many of their metastases have been treated by surgical resections; this treatment may be repeated multiple times in individuals who develop repeated local recurrences and/or metastases. Radiation therapy combined with partial surgical resection or radiation therapy alone have been used in cases where the primary, recurrent, or metastatic tumor cannot be safely or totally removed.[7][10][6] However, the experiences with radiation therapy and chemotherapy to treat LGFMS are limited and this tumor does not appear very sensitive to either treatment modality.[5] In one long-term study of 33 treated individuals with LFFMS, 14 died of their tumors from 3 to 42 years (median: 15 years) after their initial diagnosis and 19 were alive after 5½ to 70 years (median: 13 years] of their diagnosis with 6 individuals having persistent tumors and 13 being tumor-free.[6]
References
- Evans HL (November 1987). "Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having a deceptively benign appearance". American Journal of Clinical Pathology. 88 (5): 615–9. doi:10.1093/ajcp/88.5.615. PMID 3673943.
- Flucke U, van Noesel MM, Siozopoulou V, Creytens D, Tops BB, van Gorp JM, Hiemcke-Jiwa LS (June 2021). "EWSR1-The Most Common Rearranged Gene in Soft Tissue Lesions, Which Also Occurs in Different Bone Lesions: An Updated Review". Diagnostics (Basel, Switzerland). 11 (6): 1093. doi:10.3390/diagnostics11061093. PMC 8232650. PMID 34203801.
- Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- Porrino J, Al-Dasuqi K, Irshaid L, Wang A, Kani K, Haims A, Maloney E (June 2021). "Update of pediatric soft tissue tumors with review of conventional MRI appearance-part 1: tumor-like lesions, adipocytic tumors, fibroblastic and myofibroblastic tumors, and perivascular tumors". Skeletal Radiology. doi:10.1007/s00256-021-03836-2. PMID 34191084. S2CID 235678096.
- Martínez-Trufero J, Cruz Jurado J, Gómez-Mateo MC, Bernabeu D, Floría LJ, Lavernia J, Sebio A, García Del Muro X, Álvarez R, Correa R, Hernández-León CN, Marquina G, Hindi N, Redondo A, Martínez V, Asencio JM, Mata C, Valverde Morales CM, Martin-Broto J (September 2021). "Uncommon and peculiar soft tissue sarcomas: Multidisciplinary review and practical recommendations for diagnosis and treatment. Spanish group for Sarcoma research (GEIS - GROUP). Part I". Cancer Treatment Reviews. 99: 102259. doi:10.1016/j.ctrv.2021.102259. PMID 34311246.
- Evans, Harry L (2011). "Low-Grade Fibromyxoid Sarcoma: A Clinicopathologic Study of 33 Cases With Long-Term Follow-Up". The American Journal of Surgical Pathology. 35 (10): 1450–1462. doi:10.1097/PAS.0b013e31822b3687. PMID 21921785. S2CID 21592343.
- Yue Y, Liu Y, Song L, Chen X, Wang Y, Wang Z (February 2018). "MRI findings of low-grade fibromyxoid sarcoma: a case report and literature review". BMC Musculoskeletal Disorders. 19 (1): 65. doi:10.1186/s12891-018-1976-z. PMC 6389061. PMID 29482535.
- Mustafa S, VandenBussche CJ, Ali SZ, Siddiqui MT, Wakely PE (2020). "Cytomorphologic findings of low-grade fibromyxoid sarcoma". Journal of the American Society of Cytopathology. 9 (3): 191–201. doi:10.1016/j.jasc.2020.01.006. PMID 32197967. S2CID 212810533.
- Murshed KA, Al-Bozom I, Ammar A (August 2021). "Sclerosing epithelioid fibrosarcoma: in-depth review of a genetically heterogeneous tumor". APMIS. 129 (8): 455–460. doi:10.1111/apm.13157. PMID 34048081. S2CID 235232220.
- Cowan ML, Thompson LD, Leon ME, Bishop JA (June 2016). "Low-Grade Fibromyxoid Sarcoma of the Head and Neck: A Clinicopathologic Series and Review of the Literature". Head and Neck Pathology. 10 (2): 161–6. doi:10.1007/s12105-015-0647-8. PMC 4838961. PMID 26276044.
- Saab-Chalhoub MW, Al-Rohil RN (April 2019). "Low-grade fibromyxoid sarcoma of acral sites: Case report and literature review". Journal of Cutaneous Pathology. 46 (4): 271–276. doi:10.1111/cup.13413. PMID 30632203. S2CID 58592480.
- Sajid MI, Arshad S, Abdul-Ghafar J, Fatimi SH, Din NU (February 2021). "Low-grade fibromyxoid sarcoma incidentally discovered as an asymptomatic mediastinal mass: a case report and review of the literature". Journal of Medical Case Reports. 15 (1): 50. doi:10.1186/s13256-020-02605-4. PMC 7851906. PMID 33526082.
- Geramizadeh B, Zare Z, Dehghanian AR, Bolandparvaz S, Marzban M (2018). "Huge mesenteric low-grade fibromyxoid sarcoma: A case report and review of the literature". Rare Tumors. 10: 2036361318777031. doi:10.1177/2036361318777031. PMC 5971385. PMID 29854356.
- Sargar K, Kao SC, Spunt SL, Hawkins DS, Parham DM, Coffin C, McCarville MB (August 2015). "MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332". AJR. American Journal of Roentgenology. 205 (2): 414–20. doi:10.2214/AJR.14.13972. PMC 4570741. PMID 26204295.
- "CREB3L2 cAMP responsive element binding protein 3 like 2 [Homo sapiens (Human)] - Gene - NCBI".
- Sambri A, Righi A, Tuzzato G, Donati D, Bianchi G (2018). "Low-grade fibromyxoid sarcoma of the extremities: a clinicopathologic study of 24 cases and review of the literature". Polish Journal of Pathology. 69 (3): 219–225. doi:10.5114/pjp.2018.79541. hdl:11585/667908. PMID 30509048.
- Lindén M, Thomsen C, Grundevik P, Jonasson E, Andersson D, Runnberg R, Dolatabadi S, Vannas C, Luna Santamaria M, Fagman H, Ståhlberg A, Åman P (May 2019). "FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex". EMBO Reports. 20 (5). doi:10.15252/embr.201845766. PMC 6500973. PMID 30962207.
- Krystel-Whittemore M, Taylor MS, Rivera M, Lennerz JK, Le LP, Dias-Santagata D, Iafrate AJ, Deshpande V, Chebib I, Nielsen GP, Wu CL, Nardi V (November 2019). "Novel and established EWSR1 gene fusions and associations identified by next-generation sequencing and fluorescence in-situ hybridization". Human Pathology. 93: 65–73. doi:10.1016/j.humpath.2019.08.006. PMID 31430493. S2CID 201117873.
- Agaimy A (January 2020). "What is new in epithelioid soft tissue tumors?". Virchows Archiv. 476 (1): 81–96. doi:10.1007/s00428-019-02677-8. PMID 31686193. S2CID 207893952.
- Warmke LM, Meis JM (March 2021). "Sclerosing Epithelioid Fibrosarcoma: A Distinct Sarcoma With Aggressive Features". The American Journal of Surgical Pathology. 45 (3): 317–328. doi:10.1097/PAS.0000000000001559. PMID 32769431. S2CID 221084763.
- Chew W, Benson C, Thway K, Hayes A, Miah A, Zaidi S, Lee AT, Messiou C, Fisher C, van der Graaf WT, Jones RL (September 2018). "Clinical Characteristics and efficacy of chemotherapy in sclerosing epithelioid fibrosarcoma". Medical Oncology (Northwood, London, England). 35 (11): 138. doi:10.1007/s12032-018-1192-6. PMC 6132781. PMID 30187231.)
- Porteus C, Gan Q, Gong Y, Pantanowitz L, Henderson-Jackson E, Saeed-Vafa D, Mela N, Peterson D, Ahmad N, Ahmed A, Bui M (2020). "Sclerosing epithelioid fibrosarcoma: cytologic characterization with histologic, immunohistologic, molecular, and clinical correlation of 8 cases". Journal of the American Society of Cytopathology. 9 (6): 513–519. doi:10.1016/j.jasc.2020.05.005. PMID 32624384. S2CID 220369922.