Mitochondrial biogenesis
Mitochondrial biogenesis is the process by which cells increase mitochondrial numbers.[1][2] It was first described by John Holloszy in the 1960s, when it was discovered that physical endurance training induced higher mitochondrial content levels, leading to greater glucose uptake by muscles.[3] Mitochondrial biogenesis is activated by numerous different signals during times of cellular stress or in response to environmental stimuli, such as aerobic exercise.[1][2][4]
Background
The ability for a mitochondrion to self-replicate is rooted in its evolutionary history. It is commonly thought that mitochondria descend from cells that formed endosymbiotic relationships with α-protobacteria; they have their own genome for replication.[5] However, recent evidence suggests that mitochondria may have evolved without symbiosis.[6] The mitochondrion is a key regulator of the metabolic activity of the cell, and is also an important organelle in both production and degradation of free radicals.[7] It is postulated that higher mitochondrial copy number (or higher mitochondrial mass) is protective for the cell.
Mitochondria are produced from the transcription and translation of genes both in the nuclear genome and in the mitochondrial genome. The majority of mitochondrial protein comes from the nuclear genome, while the mitochondrial genome encodes parts of the electron transport chain along with mitochondrial rRNA and tRNA. Mitochondrial biogenesis increases metabolic enzymes for glycolysis, oxidative phosphorylation and ultimately a greater mitochondrial metabolic capacity. However, depending on the energy substrates available and the redox state of the cell, the cell may increase or decrease the number and size of mitochondria.[8] Critically, mitochondrial numbers and morphology vary according to cell type and context-specific demand, whereby the balance between mitochondrial fusion/fission regulates mitochondrial distribution, morphology, and function.[9][8]
Protein import
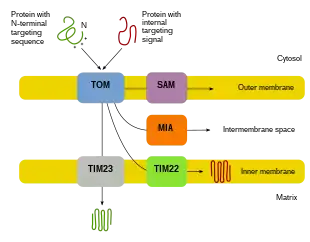
Since the majority of mitochondrial protein comes from the nuclear genome, the proteins need to be properly targeted and transported into the mitochondria to perform their functions.[8][10][11] First, mRNA is translated in the cell’s cytosol.[10][11] The resulting unfolded precursor proteins will then be able to reach their respective mitochondrial compartments.[11][10] Precursor proteins will be transported to one of four areas of the mitochondria, which include the outer membrane, inner membrane, intermembrane space, and matrix.[10][11] All proteins will enter the mitochondria by a translocase on the outer mitochondrial membrane (TOM).[11][10][5] Some proteins will have an N-terminal targeting signal, and these proteins will be detected and transported into the matrix, where they will then be cleaved and folded.[12][11][10] Other proteins may have targeting information in their sequences and will not include an N-terminal signal.[11][10] During the past two decades, researchers have discovered over thirty proteins that participate in mitochondrial protein import.[11] As researchers learn more about these proteins and how they reach the respective mitochondrial compartments that utilize them, it becomes evident that there is a multitude of processes that work together in the cell to allow for mitochondrial biogenesis.[11][8]
Fusion and fission
Mitochondria are highly versatile and are able to change their shape through fission and fusion events.[9][8] Definitively, fission is the event of a single entity breaking apart, whereas fusion is the event of two or more entities joining to form a whole.[8] The processes of fission and fusion oppose each other and allow the mitochondrial network to constantly remodel itself.[9][8] If a stimulus induces a change in the balance of fission and fusion in a cell, it could significantly alter the mitochondrial network.[9][13] For example, an increase in mitochondrial fission would create many fragmented mitochondria, which has been shown to be useful for eliminating damaged mitochondria and for creating smaller mitochondria for efficient transporting to energy-demanding areas.[13][14] Therefore, achieving a balance between these mechanisms allows a cell to have the proper organization of its mitochondrial network during biogenesis and may have an important role in muscle adaptation to physiological stress.[13]
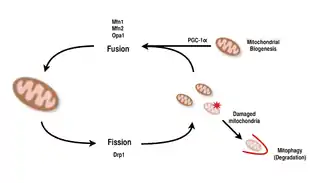
In mammals, mitochondrial fusion and fission are both controlled by GTPases of the dynamin family.[8][13] The process of mitochondrial fission is directed by Drp1, a member of the cytosolic dynamin family.[8][9] This protein forms a spiral around mitochondria and constricts to break apart both the outer and inner membranes of the organelle.[14] On the other hand, the process of fusion is directed by different membrane-anchored dynamin proteins at different levels of the mitochondria.[13] Fusion at the level of the outer mitochondrial membrane is mediated by Mfn1 and Mfn2 (Mitofusins 1 and 2),[15] and fusion at the level of the inner mitochondrial membrane is mediated by Opa1.[8][12][13] Multiple research studies have observed correlated increases between mitochondrial respiratory capacity with Mfn1, Mnf2, and Drp1 gene expression after endurance exercises.[14][15] Therefore, it is supported that reorganization of the mitochondrial network in muscle cells plays an important role in response to exercise.[4][13][15]
Regulation
PGC-1α, a member of the peroxisome proliferator-activated receptor gamma (PGC) family of transcriptional coactivators, is the master regulator of mitochondrial biogenesis.[1][2][16] It is known to co-activate nuclear respiratory factor 2 (NRF2/GABPA), and together with NRF-2 coactivates nuclear respiratory factor 1 (NRF1).[15][16] The NRFs, in turn, activate the mitochondrial transcription factor A (tfam), which is directly responsible for transcribing nuclear-encoded mitochondrial proteins.[15][16] This includes both structural mitochondrial proteins as well as those involved in mtDNA transcription, translation, and repair.[16] PGC- 1β, a protein that is structurally similar to PGC-1α, is also involved in regulating mitochondrial biogenesis, but differs in that it does not get increased in response to exercise.[5][17][16] While there have been significant increases in mitochondria found in tissues where PGC-1α is overexpressed, as the cofactor interacts with these key transcription factors, knockout mice with disrupted PGC-1α are still viable and show normal mitochondrial abundance.[17][5][16] Thus, PGC-1α is not required for normal development of mitochondria in mice, but when put under physiological stress, these mice exhibit diminished tolerance compared to mice with normal levels of PGC-1α.[5][16][17] Similarly, in knockout mice with disrupted PGC-1β, the mice showed mostly normal levels of mitochondrial function with decreased ability to adapt to physiological stress.[18][5] However, a double knockout experiment of PGC-1α/β created mice that died mostly within 24 hours by defects in mitochondrial maturation of cardiac tissue.[19] These findings suggest that while both PGC-1α and PGC- 1β do not each solely establish a cell’s ability to perform mitochondrial biogenesis, together they are able to complement each other for optimal mitochondrial maturation and function during periods of physiological stress.[19][5][17]
AMP-activated kinase (AMPK) also regulates mitochondrial biogenesis by phosphorylating and activating PGC-1α upon sensing an energy deficiency in muscle.[5][16] In mice with reduced ATP/AMP ratios that would occur during exercise, the energy depletion has been shown to correlate with AMPK activation.[5][18][16] AMPK activation then continued to activate PGC- 1α and NRFs in these mice, and mitochondrial biogenesis was stimulated.[5][18][16]
Aging
The capacity for mitochondrial biogenesis has been shown to decrease with age, and such decreased mitochondrial function has been associated with diabetes and cardiovascular disease.[20][21][22] Aging and disease can induce changes in the expression levels of proteins involved in the fission and fusion mechanisms of mitochondria, thus creating dysfunctional mitochondria.[23][24] One hypothesis for the detrimental results of aging is associated with the loss of telomeres, the end segments of chromosomes that protect genetic information from degradation.[21][24] Telomere loss has also been associated with decreased mitochondrial function.[24][21] Deficiency of telomerase reverse transcriptase (TERT), an enzyme that plays a role in preserving telomeres, has been correlated with activated p53, a protein that suppresses PGC-1α.[24][23][21] Therefore, the loss of telomeres and TERT that comes with aging has been associated with impaired mitochondrial biogenesis.[21][23][24] AMPK expression has also been shown to diminish with age, which may also contribute to suppressing mitochondrial biogenesis.[5][24]
Clinical Applications of Targeting Mitochondrial Biogenesis
Mitochondrial biogenesis can be targeted to prevent cancer proliferation. Specifically, two biogenesis regulators—PGC1α and c-Myc—can be targeted to prevent cancer proliferation. PGC1α is a key component in mitochondrial biogenesis—as a transcriptional coactivator, it targets multiple transcription factors and the estrogen-related receptor alpha (ERRα).[25] Compounds that target the pathway between PGC1α and ERRα, such as the ERRα inverse agonist, XCT-790, have been found to significantly decrease mitochondrial biogenesis, thus greatly reducing cancer cells’ proliferation and increasing their sensitivity to chemotherapeutic agents.[26] c-Myc, a transcription factor, can be inhibited during its dimerization with Max protein by molecules such as IIA6B17[27] and omomyc.[28] Inhibition of the c-Myc-Max complex can block the cell cycle and induce apoptosis in cancer cells.
References
- Valero T (2014). "Editorial (Thematic Issue: Mitochondrial Biogenesis: Pharmacological Approaches)". Current Pharmaceutical Design. 20 (35): 5507–5509. doi:10.2174/138161282035140911142118. hdl:10454/13341. PMID 24606795.
- Sanchis-Gomar F, García-Giménez JL, Gómez-Cabrera MC, Pallardó FV (2014). "Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches". Current Pharmaceutical Design. 20 (35): 5619–33. doi:10.2174/1381612820666140306095106. PMID 24606801.
- Holloszy JO (April 2011). "RRegulation of Mitochondrial Biogenesis and GLUT4 Expression by Exercise". Comprehensive Physiology. 1 (2): 921–40. doi:10.1002/cphy.c100052. ISBN 9780470650714. PMID 23737207.
- Boushel R, Lundby C, Qvortrup K, Sahlin K (October 2014). "Mitochondrial plasticity with exercise training and extreme environments". Exercise and Sport Sciences Reviews. 42 (4): 169–74. doi:10.1249/JES.0000000000000025. PMID 25062000.
- Jornayvaz FR, Shulman GI (2010). "Regulation of mitochondrial biogenesis". Essays in Biochemistry. 47: 69–84. doi:10.1042/bse0470069. PMC 3883043. PMID 20533901.
- Harish A, Kurland CG (December 2017). "Mitochondria are not captive bacteria". Journal of Theoretical Biology. 434: 88–98. doi:10.1016/j.jtbi.2017.07.011. PMID 28754286.
- Bevilacqua L, Ramsey JJ, Hagopian K, Weindruch R, Harper ME (May 2004). "Effects of short- and medium-term calorie restriction on muscle mitochondrial proton leak and reactive oxygen species production". American Journal of Physiology. Endocrinology and Metabolism. 286 (5): E852-61. doi:10.1152/ajpendo.00367.2003. PMID 14736705.
- Mishra P, Chan DC (February 2016). "Metabolic regulation of mitochondrial dynamics". The Journal of Cell Biology. 212 (4): 379–87. doi:10.1083/jcb.201511036. PMC 4754720. PMID 26858267.
- Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, et al. (June 2016). "Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity". Neurobiology of Disease. 90: 3–19. doi:10.1016/j.nbd.2015.10.011. PMID 26494254.
- Dudek J, Rehling P, van der Laan M (February 2013). "Mitochondrial protein import: common principles and physiological networks". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1833 (2): 274–85. doi:10.1016/j.bbamcr.2012.05.028. PMID 22683763.
- Baker MJ, Frazier AE, Gulbis JM, Ryan MT (September 2007). "Mitochondrial protein-import machinery: correlating structure with function". Trends in Cell Biology. 17 (9): 456–64. doi:10.1016/j.tcb.2007.07.010. PMID 17825565.
- Ventura-Clapier R, Garnier A, Veksler V (July 2008). "Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha". Cardiovascular Research. 79 (2): 208–17. doi:10.1093/cvr/cvn098. PMID 18430751.
- Youle RJ, van der Bliek AM (August 2012). "Mitochondrial fission, fusion, and stress". Science. 337 (6098): 1062–5. doi:10.1126/science.1219855. PMC 4762028. PMID 22936770.
- Bo H, Zhang Y, Ji LL (July 2010). "Redefining the role of mitochondria in exercise: a dynamic remodeling". Annals of the New York Academy of Sciences. 1201: 121–8. doi:10.1111/j.1749-6632.2010.05618.x. PMID 20649548.
- Cartoni R, Léger B, Hock MB, Praz M, Crettenand A, Pich S, et al. (August 2005). "Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise". The Journal of Physiology. 567 (Pt 1): 349–58. doi:10.1113/jphysiol.2005.092031. PMC 1474174. PMID 15961417.
- Johri A, Chandra A, Flint Beal M (September 2013). "PGC-1α, mitochondrial dysfunction, and Huntington's disease". Free Radical Biology & Medicine. 62: 37–46. doi:10.1016/j.freeradbiomed.2013.04.016. PMC 3722269. PMID 23602910.
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, et al. (October 2004). "Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice". Cell. 119 (1): 121–35. doi:10.1016/j.cell.2004.09.013. PMID 15454086.
- Scarpulla RC (July 2011). "Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1813 (7): 1269–78. doi:10.1016/j.bbamcr.2010.09.019. PMC 3035754. PMID 20933024.
- Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, et al. (July 2008). "Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart". Genes & Development. 22 (14): 1948–61. doi:10.1101/gad.1661708. PMC 2492740. PMID 18628400.
- Handy DE, Loscalzo J (June 2012). "Redox regulation of mitochondrial function". Antioxidants & Redox Signaling. 16 (11): 1323–67. doi:10.1089/ars.2011.4123. PMC 3324814. PMID 22146081.
- David R (April 2011). "Ageing: Mitochondria and telomeres come together". Nature Reviews. Molecular Cell Biology. 12 (4): 204. doi:10.1038/nrm3082. PMID 21407239.
- Hagen TM, Wehr CM, Ames BN (November 1998). "Mitochondrial decay in aging. Reversal through supplementation of acetyl-L-carnitine and N-tert-butyl-alpha-phenyl-nitrone". Annals of the New York Academy of Sciences. 854: 214–23. doi:10.1111/j.1749-6632.1998.tb09904.x. PMID 9928432.
- Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, et al. (February 2011). "Telomere dysfunction induces metabolic and mitochondrial compromise". Nature. 470 (7334): 359–65. doi:10.1038/nature09787. PMC 3741661. PMID 21307849.
- Sahin E, DePinho RA (May 2012). "Axis of ageing: telomeres, p53 and mitochondria". Nature Reviews. Molecular Cell Biology. 13 (6): 397–404. doi:10.1038/nrm3352. PMC 3718675. PMID 22588366.
- Scarpulla, Richard (2011). "Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network". Biochim Biophys Acta. 1813 (7). doi:10.1016/j.bbamcr.2010.09.019.
- Kokabu, Tetsuya; Mori, Taisuke; Matsushima, Hiroshi; Yoriki, Kaori; Kataoka, Hisashi; Tarumi, Yosuke; Kitawaki, Jo (2019). "Antitumor effect of XCT790, an ERRα inverse agonist, on ERα-negative endometrial cancer cells". Cell Oncol (Dordr). 42 (2). doi:10.1007/s13402-019-00423-5.
- Lu, Xiaohong; Vogt, Peter; Boger, Dale; Lunec, John (2008). "Disruption of the MYC transcriptional function by a small-molecule antagonist of MYC/MAX dimerization". Oncol. Rep. 19 (3). PMID 18288422.
- Demma, Mark; Mapelli, Claudio; Sun, Angie; Bodea, Smaranda; Ruprecht, Benjamin; Javaid, Sarah; Wiswell, Derek; Muise, Eric; Chen, Shiyang; Zelina, John; Orvieto, Federica; Santoprete, Alessia; Altezza, Simona; Tucci, Federica; Escandon, Enrique; Hall, Brian; Ray, Kallol; Walji, Abbas; O'Neil, Jennifer (2019). "Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene". Mol Cell Biol. 39 (22). doi:10.1128/MCB.00248-19. PMC 6817756.
Further reading
- Smith JA, Stallons LJ, Collier JB, Chavin KD, Schnellmann RG (February 2015). "Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury". The Journal of Pharmacology and Experimental Therapeutics. 352 (2): 346–57. doi:10.1124/jpet.114.221085. PMC 4293437. PMID 25503387.
- Cameron RB, Beeson CC, Schnellmann RG (December 2016). "Development of Therapeutics That Induce Mitochondrial Biogenesis for the Treatment of Acute and Chronic Degenerative Diseases". Journal of Medicinal Chemistry. 59 (23): 10411–10434. doi:10.1021/acs.jmedchem.6b00669. PMC 5564430. PMID 27560192.
- Whitaker RM, Corum D, Beeson CC, Schnellmann RG (2016). "Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases". Annual Review of Pharmacology and Toxicology. 56: 229–49. doi:10.1146/annurev-pharmtox-010715-103155. PMID 26566156.