Myoclonic dystonia
Myoclonic dystonia or Myoclonus dystonia syndrome is a rare movement disorder that induces spontaneous muscle contraction causing abnormal posture. The prevalence of myoclonus dystonia has not been reported, however, this disorder falls under the umbrella of movement disorders which affect thousands worldwide.[1] Myoclonus dystonia results from mutations in the SGCE gene coding for an integral membrane protein found in both neurons and muscle fibers. Those suffering from this disease exhibit symptoms of rapid, jerky movements of the upper limbs (myoclonus), as well as distortion of the body's orientation due to simultaneous activation of agonist and antagonist muscles (dystonia).
| Myoclonus dystonia |
|---|
Myoclonus dystonia is caused by loss-of-function-mutations in the epsilon sarcoglycan gene (SGCE). The disease is dominantly inherited, however SGCE is an imprinted gene,[2] so only the paternal allele is expressed. Therefore, children suffering from this disease inherit the mutation from the father. If the mutated allele is inherited from the mother, the child is not likely to exhibit symptoms.
While no cure has been found for myoclonus dystonia, treatment options are available to those suffering from the disease. Ethanol often ameliorates the symptoms well, and so the syndrome is also called "Alcohol-responsive dystonia". Alcohol may be substituted by benzodiazepines, such as clonazepam, which work through the same mechanism. Deep brain stimulation (DBS) is another viable option that can alleviate symptoms without the unwanted side effects of medications, and has been successful in treating other movement disorders.[3]
Signs and symptoms
Myoclonus dystonia is characterized by two primary features: myoclonus and dystonia. For the majority of individuals with myoclonus dystonia, the myoclonus component of the disorder is often the primary and most disabling feature in comparison to the dystonia component. The symptoms of myoclonus dystonia vary substantially in severity.
Myoclonus
Myoclonus is characterized by rapid contractions that affect the upper body including the neck, torso and arms, but may also affect the legs. These movements are stimulated by various factors including stress, noise, caffeine, and physical stimuli. Myoclonus can be characterized in multiple ways including neurological basis, muscular activity, and by stimuli. Myoclonus can be positive or negative; positive myoclonus results from brief spurts of muscle activity and negative myoclonus occurs when there is a lack of any muscular activity. Myoclonus is usually classified physiologically to optimize treatment. Myoclonus is a precursor effect to myoclonus dystonia and most commonly begins in childhood or adolescence.[4][5]
Myoclonus is classified as cortical, subcortical, peripheral or spinal. Cortical myoclonus is the most common of these four and affects the upper limbs and face. Myoclonus dystonia has been characterized under subcortical origin, specifically under nonsegmented myoclonus or brainstem myoclonus. Symptoms within this classification include the startle response and reticular reflex myoclonus. Sudden stimuli like noise or touch to areas around the head or chest cause the startle response which will go up the brain stem and down the spinal cord causing jerk-like movements. Hyperekplexia is a heightened brainstem response where an affected person will continue to elicit the same response to a repeated stimuli. In contrast, reticular reflex myoclonus occurs spontaneously to stimuli applied to distal limbs. Spinal myoclonus is caused by defects in spinal organization or connections, and peripheral myoclonus has symptoms of rhythmic jerks due to a neuron-the most common being the hemifacial spasm.[5]
Dystonia
Dystonia is a response to simultaneous contraction of agonist and antagonist muscles seen as twisting and contorting that affect posture and stance. Other symptoms can include tremors and muscle spasms due to various interactions of muscle, contractions and movement.[4] Dystonia can be either primary or secondary with the latter being more common. Primary dystonia or "pure" dystonia is only physiological in origin. Secondary dystonia has multiple origins that are physiological, pathological or neurological.[6][7]
Myoclonus dystonia
Myoclonus dystonia includes the rapid contractions of myoclonus alongside the abnormal postures classified under dystonia, as well as neurological and psychiatric issues. This disease typically begins during childhood with symptoms of myoclonus and slight dystonia, most commonly cervical dystonia or writer's cramp. Dystonia symptoms tend to not get exaggerated over the course of the disease and is rarely the only associated symptom, while the myoclonus symptoms can become more severe. Psychiatric issues are clinically diagnosed with the aforementioned symptoms and include depression, anxiety, personality disorders and addiction. Obsessive-compulsive disorder is associated with myoclonus dystonia as both have been found to have a commonality on chromosome 7 in various studies.[4]
Neurological symptoms are relatively common in those with myoclonus dystonia. Any neurological abnormalities will not normally be present in those affected at a young age. Neurological testing has been performed to determine the origins of these symptoms and multiple parts of the brain have been pinpointed including the brainstem, neocortex, pallidum, and thalamus. These cause various effects in those diagnosed with myoclonus dystonia including changes in posture and tremors, and very rarely dementia and ataxia.[4]
Cause
The majority of myoclonus dystonia cases are the result of a mutation in the epsilon sarcoglycan gene (SGCE). This gene is found on chromosome 7, with its specific cytogenic location being 7q21.3. The 70,985 bp SGCE gene encodes the protein epsilon (ε)-sarcoglycan. The five proteins that make up the sarcoglycan family function as integral membrane proteins that anchor the cytoskeleton of cells to the extracellular matrix. Epsilon sarcoglycan is a membrane protein that can be found in the liver, lungs, kidney, and spleen, but is most prevalent in muscle and neuronal cells. Its prevalence in both muscle fibers and the synapses of neurons suggest why symptoms of both myoclonus and dystonia appear from the improperly functioning protein. Recessive mutations in the other sarcoglycans also result in muscular disorders, further supporting that mutations in the SGCE gene cause myoclonus dystonia.[8]
Epsilon sarcoglycan itself is part of the dystrophin-associated protein (DAP) complex that binds the sarcolemma of muscle cells to the extracellular connective tissue. The purpose is to reduce the mechanical force on the sarcolemma as a result of muscle contraction. In addition to myoclonus dystonia, problems associated with a dysfunctional DAP complex include Duchenne muscular dystrophy.[9]
Upwards of 65 mutations of the SGCE gene are thought to cause myoclonus dystonia. The majority of the mutations lead to a truncated protein product that results in the loss-of-function of the epsilon sarcoglycan protein.[10] The dysfunctional protein is ultimately recycled by the cell by degradation mediated by the proteasome, resulting in significant shortages of the integral membrane protein in both neurons and muscle fibers.
The mutant allele is inherited in a dominant fashion—that is the mutation can be inherited if one parent has that allele. However, genomic imprinting occurs on the mother's allele, so only the father's allele is expressed.[2][10] Therefore, inheriting a mutated, paternal allele of the SGCE gene will result in the expression of the dysfunctional epsilon sarcoglycan protein. Offspring will not produce a mutant protein product in 95% of cases where the mother passes on a mutation in the SGCE gene.[10]
While SGCE gene mutations are the central cause of myoclonus dystonia, there have been separate cases where individuals and families present symptoms akin to myoclonus dystonia but lack the mutations at this locus. Base-pair deletions of the DYT1 gene, missense mutations in the DRD2 gene, maternal uniparental disomy, and chromosome 18 linkage have all been associated in rare cases myoclonus dystonia where the SGCE gene is unaffected.[4]
Treatment
To date, there is no single, universal treatment that has been found to cure myoclonus dystonia. However, there are several treatment methods that have been found to be effective for helping to reduce the symptoms associated with the syndrome.
Medications
Many drugs used to treat myoclonus dystonia do not have a significant impact individually, but when combined, can work on different brain mechanisms to best alleviate symptoms. The method of treatment used depends on the severity of the symptoms presented in the individual, and whether the underlying cause of the syndrome is known.
Benzodiazepines
Benzodiazepines such as clonazepam improve tremors caused by the myoclonus aspect of this syndrome by binding allosterically to GABAA ionotropic receptors, causing an influx of chloride ions that produce an inhibitory effect that can calm myoclonic jerks.[4][11]
Antiepileptics
Antiepileptics like valproate must act upon GABA receptors and manipulate ionic conductance to reduce tremors and spasms in myoclonus dystonia. GABA neurons that fire rapidly and affect the motor cortex are blocked by antiepileptics in addition to changes in sodium and calcium concentrations that can excite the neuron. Different antiepileptics vary in sufficiency to control ionic conductance and can also produce seizures or myoclonus symptoms in some patients.[12] Another agent that has been used is zonisamide.[13]
Anticholinergics
Anticholinergics like benzatropine alleviate dystonia symptoms by blocking the activity of acetylcholine. Acetylcholine is involved in the pathophysiology of dystonia within the basal ganglia, although its exact role has not been determined. Acetylcholine is involved with dopamine and glutamate pathways in the basal ganglia, in addition to presynaptic muscarinic receptors which are involved in motor control. Acetylcholine is usually overactive in dystonia patients and blocking of this neurotransmitter would reduce contortion of the upper body, but can produce side effects of drowsiness, confusion and memory issues in adults.[14]
Botulinum toxin
Botulinum toxin injections also act upon acetylcholine to reduce dystonia symptoms. The neurotoxin is active in presynaptic terminals and blocks exocytosis of acetylcholine into the synaptic cleft which reduces muscle activity. Botulinum may also have a role in inhibiting glutamate and changing muscle movement. Studies have also shown possible axon transport of this neurotoxin as well as its function as a pain reliever without effect on overactive muscle movement in myoclonus dystonia patients.[15]
Alcohol
Consumption of alcohol has also been found to be an effective agent for temporarily easing the severity of the tremors associated with myoclonus dystonia. Alcohol causes an increase in GABA transmission between interneurons and Purkinje cells. This then reduces the transmission of glutamate at granule cell-Purkinje cell synapses, which decreases muscle movements. This treatment only alleviates the strength of the tremors for a short duration and does not change how often tremors will occur. Doctors inform patients of risks associated with the use of alcohol for myoclonus dystonia due to the high susceptibility for excessive alcohol use and dependency. Alcohol use disorder itself causes tremors in the hands and degeneration of the Purkinje cells and other parts of the cerebral cortex, counteracting alcohol's original corrective effects.[16]
Deep brain stimulation
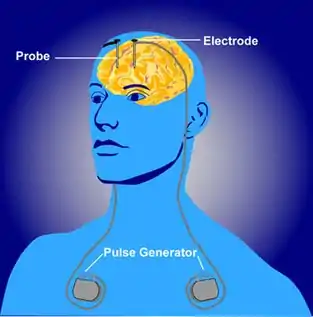
Deep brain stimulation (DBS) has been found to be an effective and safe treatment for myoclonus dystonia patients, whose severe and debilitating symptoms are resistant to drug treatments. Electrical stimulation within the brain is a common treatment for many movement disorders because of the ability to excite or inhibit neurons within the brain. Deep brain stimulation patients have electrodes inserted into the brain and then an electrical signal is sent from an external source to elicit a response. The frequency and intensity of this signal can be changed to monitor the effects on neuronal activity using voltage recordings or neuroimaging, like functional MRIs. By re-positioning the electrodes in different areas or changing the size or timing of the stimulus, varying effects can be seen on the patient depending on the origin of the disorder.[3]
In one study, five patients with genetically determined epsilon sarcoglycan protein deficiency underwent deep brain stimulation of the internal pallidum. Each patient's movement and disability symptoms were assessed before and after treatment using the Burke-Fahn-Marsden Dystonia Rating Scale and the Unified Myoclonus Rating Scale. Upon completion of the surgery, both the myoclonus and dystonia symptoms of the disorder had decreased by 70%, with no report of unfavorable side effects. Therefore, deep brain stimulation has been shown to effectively improve both myoclonus and dystonia, unlike many drug treatments which may improve one or the other.[17]
Other studies examined the effects of DBS to both the ventrointermediate nucleus of the thalamus, Vim, and the globus pallidus interna, GPi. Following deep brain stimulation of GPi and Vim, the Unified Myoclonus Rating Scale disability score improved 61-66%. In addition, the Dystomia Rating Scale score improved by 45-48%. While there was no significant difference in improvement between GPi-Vim stimulation and GPi stimulation, GPi-Vim stimulation was significantly more effective than Vim deep brain stimulation alone. Overall, Deep brain stimulation shows promise as a viable treatment for myoclonus dystonia.[18]
Although myoclonus and dystonia are present in myoclonus dystonia patients, optimum treatment for myoclonus dystonia differs from the treatment for myoclonus or dystonia alone. Myoclonus improved significantly more than dystonia when Deep brain stimulation was applied. In addition, myoclonus improved regardless of whether Deep brain stimulation was applied to GPi or Vim. However, GPi stimulation was more effective at reducing the symptoms of dystonia than Vim stimulation.[17]
References
- Wenning, Gregor K.; Kiechl, Stefan; Seppi, Klaus; Müller, Joerg; Högl, Birgit; Saletu, Michael; Rungger, Gregor; Gasperi, Arno; Willeit, Johann; Poewe, Werner (1 December 2005). "Prevalence of movement disorders in men and women aged 50-89 years (Bruneck Study cohort): a population-based study". The Lancet. Neurology. 4 (12): 815–820. doi:10.1016/S1474-4422(05)70226-X. PMID 16297839. S2CID 1587090.
- Grabowski M, Zimprich A, Lorenz-Depiereux B, et al. (February 2003). "The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted". Eur. J. Hum. Genet. 11 (2): 138–44. doi:10.1038/sj.ejhg.5200938. PMID 12634861.
- Kringelbach, Morten L.; Jenkinson, Ned; Owen, Sarah L. F.; Aziz, Tipu Z. (2007-08-01). "Translational principles of deep brain stimulation". Nature Reviews Neuroscience. 8 (8): 623–635. doi:10.1038/nrn2196. ISSN 1471-003X. PMID 17637800. S2CID 147427108.
- Raymond, Deborah; Ozelius, Laurie (1993-01-01). "SGCE Myoclonus-Dystonia". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora J.H.; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (eds.). Myoclonus-Dystonia. Seattle (WA): University of Washington, Seattle. PMID 20301587.
- Kojovic, Maja; Cordivari, Carla; Bhatia, Kailash (2011-01-01). "Myoclonic disorders: a practical approach for diagnosis and treatment". Therapeutic Advances in Neurological Disorders. 4 (1): 47–62. doi:10.1177/1756285610395653. ISSN 1756-2856. PMC 3036960. PMID 21339907.
- Spatola, Marianna; Wider, Christian (2012-01-01). "Overview of primary monogenic dystonia". Parkinsonism & Related Disorders. 18 Suppl 1: S158–161. doi:10.1016/S1353-8020(11)70049-9. ISSN 1873-5126. PMID 22166420.
- Albanese, Alberto; Bhatia, Kailash; Bressman, Susan B.; DeLong, Mahlon R.; Fahn, Stanley; Fung, Victor S.C.; Hallett, Mark; Jankovic, Joseph; Jinnah, H.A. (2013-06-15). "Phenomenology and classification of dystonia: a consensus update". Movement Disorders. 28 (7): 863–873. doi:10.1002/mds.25475. ISSN 0885-3185. PMC 3729880. PMID 23649720.
- Hack, A. A.; Groh, M. E.; McNally, E. M. (2000-02-01). "Sarcoglycans in muscular dystrophy". Microscopy Research and Technique. 48 (3–4): 167–180. doi:10.1002/(SICI)1097-0029(20000201/15)48:3/4<167::AID-JEMT5>3.0.CO;2-T. ISSN 1059-910X. PMID 10679964.
- Ervasti, James M. (2013-01-01). "Structure and Function of the Dystrophin-Glycoprotein Complex". Madame Curie Bioscience Database.
- "SGCE gene". Genetics Home Reference. 2016-03-28. Retrieved 2016-04-07.
- Rudolph, Uwe; Möhler, Hanns (1 February 2006). "GABA-based therapeutic approaches: GABAA receptor subtype functions". Current Opinion in Pharmacology. 6 (1): 18–23. doi:10.1016/j.coph.2005.10.003. PMID 16376150.
- Caviness, John N. (2014-01-01). "Treatment of Myoclonus". Neurotherapeutics. 11 (1): 188–200. doi:10.1007/s13311-013-0216-3. ISSN 1933-7213. PMC 3899494. PMID 24037428.
- Salamon A, Zádori D, Horváth E, Vécsei L, Klivényi P (2019) Zonisamide treatment in myoclonus-dystonia. Orv Hetil 160(34):1353-1357
- Eskow Jaunarajs, K. L.; Bonsi, P.; Chesselet, M. F.; Standaert, D. G.; Pisani, A. (2015-04-01). "Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia". Progress in Neurobiology. 127–128: 91–107. doi:10.1016/j.pneurobio.2015.02.002. PMC 4420693. PMID 25697043.
- Oh, Hyun-Mi; Chung, Myung Eun (2015-08-14). "Botulinum Toxin for Neuropathic Pain: A Review of the Literature". Toxins. 7 (8): 3127–3154. doi:10.3390/toxins7083127. ISSN 2072-6651. PMC 4549742. PMID 26287242.
- Deik, Andres; Saunders-Pullman, Rachel; Luciano, Marta San (2012-09-01). "Substances of abuse and movement disorders: complex interactions and comorbidities". Current Drug Abuse Reviews. 5 (3): 243–253. doi:10.2174/1874473711205030243. ISSN 1874-4737. PMC 3966544. PMID 23030352.
- Azoulay-Zyss, Julie; Roze, Emmanuel; Welter, Marie-Laure; Navarro, Soledad; Yelnik, Jérôme; Clot, Fabienne; Bardinet, Eric; Karachi, Carine; Dormont, Didier; Galanaud, Damien; Pidoux, Bernard; Cornu, Philippe; Vidailhet, Marie; Grabli, David (1 January 2011). "Bilateral deep brain stimulation of the pallidum for myoclonus-dystonia due to ε-sarcoglycan mutations: A pilot study". Archives of Neurology. 68 (1): 94–98. doi:10.1001/archneurol.2010.338. PMID 21220679.
- Smith, Kara M.; Spindler, Meredith A. (2015-02-02). "Uncommon Applications of Deep Brain Stimulation in Hyperkinetic Movement Disorders". Tremor and Other Hyperkinetic Movements. 5: 278. doi:10.7916/D84X56HP. ISSN 2160-8288. PMC 4314611. PMID 25713746.