Neuroinflammation
Neuroinflammation is inflammation of the nervous tissue. It may be initiated in response to a variety of cues, including infection, traumatic brain injury,[1] toxic metabolites, or autoimmunity.[2] In the central nervous system (CNS), including the brain and spinal cord, microglia are the resident innate immune cells that are activated in response to these cues.[2] The CNS is typically an immunologically privileged site because peripheral immune cells are generally blocked by the blood–brain barrier (BBB), a specialized structure composed of astrocytes and endothelial cells.[3] However, circulating peripheral immune cells may surpass a compromised BBB and encounter neurons and glial cells expressing major histocompatibility complex molecules, perpetuating the immune response.[4] Although the response is initiated to protect the central nervous system from the infectious agent, the effect may be toxic and widespread inflammation as well as further migration of leukocytes through the blood–brain barrier.[2]
Causes
Neuroinflammation is widely regarded as chronic, as opposed to acute, inflammation of the central nervous system.[5] Acute inflammation usually follows injury to the central nervous system immediately, and is characterized by inflammatory molecules, endothelial cell activation, platelet deposition, and tissue edema.[6] Chronic inflammation is the sustained activation of glial cells and recruitment of other immune cells into the brain. It is chronic inflammation that is typically associated with neurodegenerative diseases. Common causes of chronic neuroinflammation include:
- Toxic metabolites
- Autoimmunity
- Ageing
- Microbes
- Viruses
- Traumatic brain injury
- Spinal cord injury
- Air pollution
- Passive smoke
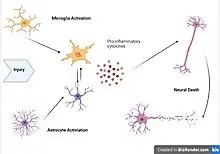
Viruses, bacteria, and other infectious agents activate the body’s defense systems and cause immune cells to protect the designed area from the damage. Some of these foreign pathogens can trigger a strong inflammatory response that can compromise the integrity of the blood-brain barrier and thus change the flow of inflammation in nearby tissue. The location along with the type of infection can determine what type of inflammatory response is activated and whether specific cytokines or immune cells will act.[7]
Neuroimmune response
Glial cells
Microglia are recognized as the innate immune cells of the central nervous system.[2] Microglia actively survey their environment and change their cell morphology significantly in response to neural injury.[8] Acute inflammation in the brain is typically characterized by rapid activation of microglia.[5] During this period, there is no peripheral immune response. Over time, however, chronic inflammation causes the degradation of tissue and of the blood–brain barrier. During this time, microglia generate reactive oxygen species and release signals to recruit peripheral immune cells for an inflammatory response.[8]
Astrocytes are glial cells that are the most abundant cells in the brain. They are involved in maintenance and support of neurons and compose a significant component of the blood–brain barrier. After insult to the brain, such as traumatic brain injury, astrocytes may become activated in response to signals released by injured neurons or activated microglia.[6][1] Once activated, astrocytes may release various growth factors and undergo morphological changes. For example, after injury, astrocytes form the glial scar composed of a proteoglycan matrix that hinders axonal regeneration.[6] However, more recent studies revealed that glia scar is not detrimental, but is in fact beneficial for axonal regeneration.[9]
Cytokines
Cytokines are a class of proteins regulating inflammation, cell signaling, and various cell processes such as growth and survival.[10] Chemokines are a subset of cytokines that regulate cell migration, such as attracting immune cells to a site of infection or injury.[10] Various cell types in the brain may produce cytokines and chemokines such as microglia, astrocytes, endothelial cells, and other glial cells. Physiologically, chemokines and cytokines function as neuromodulators that regulate inflammation and development. In the healthy brain, cells secrete cytokines to produce a local inflammatory environment to recruit microglia and clear the infection or injury. However, in neuroinflammation, cells may have sustained release of cytokines and chemokines which may compromise the blood–brain barrier.[11] Peripheral immune cells are called to the site of injury via these cytokines and may now migrate across the compromised blood brain barrier into the brain. Common cytokines produced in response to brain injury include: interleukin-6 (IL-6), which is produced during astrogliosis, and interleukin-1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α), which can induce neuronal cytotoxicity. Although the pro-inflammatory cytokines may cause cell death and secondary tissue damage, they are necessary to repair the damaged tissue.[12] For example, TNF-α causes neurotoxicity at early stages of neuroinflammation, but contributes to tissue growth at later stages of inflammation.
Peripheral immune response
The blood–brain barrier is a structure composed of endothelial cells and astrocytes that forms a barrier between the brain and circulating blood. Physiologically, this enables the brain to be protected from potentially toxic molecules and cells in the blood. Astrocytes form tight junctions, and therefore may strictly regulate what may pass the blood–brain barrier and enter the interstitial space.[6] After injury and sustained release of inflammatory factors such as chemokines, the blood–brain barrier may be compromised, becoming permeable to circulating blood components and peripheral immune cells. Cells involved in the innate and adaptive immune responses, such as macrophages, T cells, and B cells, may then enter into the brain. This exacerbates the inflammatory environment of the brain and contributes to chronic neuroinflammation and neurodegeneration.
Traumatic brain injury
Traumatic brain injury (TBI) is brain trauma caused by significant force to the head.[6] Following TBI, there are both reparative and degenerative mechanisms that lead to an inflammatory environment. Within minutes of injury, pro-inflammatory cytokines are released. The pro-inflammatory cytokine Il-1β is one such cytokine that exacerbates the tissue damage caused by TBI. TBI may cause significant damage to vital components to the brain, including the blood–brain barrier. Il-1β causes DNA fragmentation and apoptosis, and together with TNF-α may cause damage to the blood–brain barrier and infiltration of leukocytes.[13] Increased density of activated immune cells have been found in the human brain after concussion.[1]
As the most abundant immune cells in the brain, Microglia are important to the brain’s defense against injury. The major caveat of these cells comes from the fact that their ability to promote recovery mechanism with anti-inflammatory factors, is inhibited by their secondary ability to make a large amount of pro-inflammatory cytokines. This can result in sustained brain damage as anti-inflammatory factors decrease in amount when more pro-inflammatory cytokines are produced in excess by microglia. The cytokines produced by microglia, astrocytes, and other immune cells, activate glial cells further increasing the number of pro-inflammatory factors that further prevent neurological systems from recovering. The dual nature of microglia is one example of why neuroinflammation can be helpful or hurtful under specific conditions.[14]

Spinal cord injury
Spinal Cord Injury (SCI) can be divided into three separate phases. The primary or acute phase occurs from seconds to minutes after injury, the secondary phase occurs from minutes to weeks after injury, and the chronic phase occurs from months to years following injury.[15] A primary SCI is caused by spinal cord compression or transection, leading to glutamate excitotoxicity, sodium and calcium ion imbalances, and free radical damage.[16] Neurodegeneration via apoptosis and demyelination of neuronal cells causes inflammation at the injury site.[15] This leads to a secondary SCI, whose symptoms include edema, cavitation of spinal parenchyma, reactive gliosis, and potentially permanent loss of function.[15]
During the SCI induced inflammatory response, several pro-inflammatory cytokines including interleukin 1β (IL-1β), inducible Nitric Oxide Synthase (iNOS), Interferon-γ (IFN-γ), IL-6, IL-23, and tumor necrosis factor α (TNFα) are secreted, activating local microglia and attracting various immune cells such as naive bone-marrow derived macrophages.[17] These activated microglia and macrophages play a role in the pathogenesis of SCI.
Upon infiltration of the injury site's epicenter, macrophages will undergo phenotype switching from an M2 phenotype to an M1-like phenotype. The M2 phenotype is associated with anti-inflammatory factors such as IL-10, IL-4, and IL-13 and contributes to wound healing and tissue repair. However, the M1-like phenotype is associated with pro-inflammatory cytokines and reactive oxygen species that contribute to increased damage and inflammation.[18] Factors such as myelin debris, which is formed by the injury at the damage site, has been shown to induce the phenotype shift from M2 to M1.[19] A decreased population of M2 macrophages and an increased population of M1 macrophages is associated with chronic inflammation.[19] Short term inflammation is important in clearing cell debris from the site of injury, but it is this chronic, long-term inflammation that will lead to further cell death and damage radiating from the site of injury.[20]
Aging
Aging is often associated with cognitive impairment and increased propensity for developing neurodegenerative diseases, such as Alzheimer's disease.[21] Elevated inflammatory markers seemed to accelerate the brain aging process[22] In the aged brain alone, without any evident disease, there are chronically increased levels of pro-inflammatory cytokines and reduced levels of anti-inflammatory cytokines. The homeostatic imbalance between anti-inflammatory and pro-inflammatory cytokines in aging is one factor that increases the risk for neurodegenerative disease. Additionally, there is an increased number of activated microglia in aged brains, which have increased expression of major histocompatibility complex II (MHC II), ionized calcium binding adaptor-1 (IBA1), CD86, ED1 macrophage antigen, CD4, and leukocyte common antigen.[23] These activated microglia decrease the ability for neurons to undergo long term potentiation (LTP) in the hippocampus and thereby reduce the ability to form memories.[24]

As one of the major cytokines responsible for maintaining inflammatory balance, IL-6 can also be used as a biological marker to observe the correlation between age and neuroinflammation. The same levels of IL-6 observed in the brain after injury, have also been found in the elderly and indicate the potential for cognitive impairment to develop. The unnecessary upregulation of IL-6 in the elderly population is a result of dysfunctional mediation by glial cells that can lead to the priming of glial cells and result in a more sensitive neuroinflammatory response.[25]
Role in neurodegenerative disease
Alzheimer's disease
Alzheimer's disease (AD) has historically been characterized by two major hallmarks: neurofibrillary tangles and amyloid-beta plaques.[26] Neurofibrillary tangles are insoluble aggregates of tau proteins, and amyloid-beta plaques are extracellular deposits of the amyloid-beta protein. Current thinking in AD pathology goes beyond these two typical hallmarks to suggest that a significant portion of neurodegeneration in Alzheimer's is due to neuroinflammation.[26][27] Activated microglia are seen in abundance in post-mortem AD brains. Current thought is that inflammatory cytokine-activated microglia cannot phagocytose amyloid-beta, which may contribute to plaque accumulation as opposed to clearance.[28] Additionally, the inflammatory cytokine IL-1β is upregulated in AD and is associated with decreases of synaptophysin and consequent synaptic loss. Further evidence that inflammation is associated with disease progression in AD is that individuals who take non-steroidal anti-inflammatory drugs (NSAIDs) regularly have been associated with a 67% of protection against the onset of AD (relative to the placebo group) in a four-year follow-up assessment.[29] Elevated inflammatory markers showed an association with accelerated brain aging, which might explain the link to neurodegeneration in AD-related brain regions.[22]
Parkinson's disease
The leading hypothesis of Parkinson's disease progression includes neuroinflammation as a major component.[30] This hypothesis stipulates that Stage 1 of Parkinson's disease begins in the gut, as evidenced by a large number of cases that begin with constipation. The inflammatory response in the gut may play a role in alpha-synuclein (α-Syn) aggregation and misfolding, a characteristic of Parkinson's disease pathology. If there is a balance between good bacteria and bad bacteria in the gut, the bacteria may remain contained to the gut. However, dysbiosis of good bacteria and bad bacteria may cause a “leaky” gut, creating an inflammatory response. This response aids α-Syn misfolding and transfer across neurons, as the protein works its way up to the CNS. The brainstem is vulnerable to inflammation, which would explain Stage 2, including sleep disturbances and depression. In Stage 3 of the hypothesis, the inflammation affects the substantia nigra, the dopamine producing cells of the brain, beginning the characteristic motor deficits of Parkinson's disease. Stage 4 of Parkinson's disease includes deficits caused by inflammation in key regions of the brain that regulate executive function and memory. As evidence supporting this hypothesis, patients in Stage 3 (motor deficits) that are not experiencing cognitive deficits already show that there is neuroinflammation of the cortex. This suggests that neuroinflammation may be a precursor to the deficits seen in Parkinson's disease.[30]
Amyotrophic lateral sclerosis
Unlike other neurodegenerative diseases, the exact pathophysiology of amyotrophic lateral sclerosis (ALS) is still far from being fully uncovered. Several hypotheses have been proposed to explain the development and progression of this lethal disease,[31] by which neuroinflammation is one of the above. It is characterised by the activation of microglia and astrocytes, T lymphocyte infiltration, and the production of pro-inflammatory cytokines.[32] Features of neuroinflammation were observed in the brain of living ALS patients,[33] post-mortem CNS samples,[34] and mouse models of ALS.[35] Multiple evidence has described the mechanism of how microglial and astrocyte activation can promote disease progression (reviewed by [36][37]). Replacement of mSOD1 microglia and astrocytes with the wild-type forms delayed motor neuron (MN) degeneration and extended the lifespan of ALS mice.[38][39] Infiltration of T cells was reported in both early and late stages of ALS.[38][40][41] Among all T cells, CD4+ T cells has drawn the most attention by being a neuroprotective agent during MN loss.[42] T regulatory (Treg) cells is also a safeguard against neuroinflammation, demonstrated by the evidence of inverse correlation of the number of Treg cells and disease progression/ severity.[38][43] Apart from the three phenotypes discussed, peripheral macrophages/ monocytes and the complement system are also suggested to be contributed to disease pathogenesis. Activation[44] and invasion[45][46] of peripheral monocytes observed in the spinal cord of ALS patients and mice may lead to MN loss. Expression of several complement components are reported to be upregulated in the samples isolated from ALS patients[47] and transgenic rodent models.[48] Further studies are required to elucidate their roles in ALS.
Multiple sclerosis
Multiple sclerosis is the most common disabling neurological disease of young adults.[49] It is characterized by demyelination and neurodegeneration, which contribute to the common symptoms of cognitive deficits, limb weakness, and fatigue.[50] In multiple sclerosis, inflammatory cytokines disrupt the blood–brain barrier and allow for the migration of peripheral immune cells into the central nervous system. When they have migrated into the central nervous system, B cells and plasma cells produce antibodies against the myelin sheath that insulates neurons, degrading the myelin and slowing conduction in the neurons. Additionally, T cells may enter through the blood–brain barrier, be activated by local antigen presenting cells, and attack the myelin sheath. This has the same effect of degrading the myelin and slowing conduction. As in other neurodegenerative diseases, activated microglia produce inflammatory cytokines that contribute to widespread inflammation. It has been shown that inhibiting microglia decreases the severity of multiple sclerosis.[30]
Role as a therapeutic target
Drug therapy
Because neuroinflammation has been associated with a variety of neurodegenerative diseases, there is increasing interest to determine whether reducing inflammation will reverse neurodegeneration. Inhibiting inflammatory cytokines, such as IL-1β, decreases neuronal loss seen in neurodegenerative diseases. Current treatments for multiple sclerosis include interferon-B, Glatiramer acetate, and Mitoxantrone, which function by reducing or inhibiting T Cell activation, but have the side effect of systemic immunosuppression [51] In Alzheimer's disease, the use of non-steroidal anti-inflammatory drugs decreases the risk of developing the disease.[29] Current treatments for Alzheimer's disease include NSAIDs and glucocorticoids. NSAIDs function by blocking conversion of prostaglandin H2 into other prostaglandins (PGs) and thromboxane (TX). Prostoglandins and thromboxane act as inflammatory mediators and increase microvascular permeability.
Exercise
Exercise is a promising mechanism of prevention and treatment for various diseases characterized by neuroinflammation.[23] Aerobic exercise is used widely to reduce inflammation in the periphery by activating protective systems in the body that stabilize internal environment.[52] Exercise has been shown to decrease proliferation of microglia in the brain, decrease hippocampal expression of immune-related genes and reduce expression of inflammatory cytokines such as TNF-α.
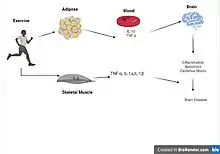
Exercise can help protect the mind and body by maintaining the brain’s internal environment, focusing on recruiting anti-inflammatory cytokines, and activating cellular processes that proactively protect against damage while also initiating recovery mechanisms. The ability of physical activity to stimulate immune defenses against neuroinflammation-related diseases has been observed in recent clinical studies. The application of various exercises under a range of different conditions resulted in higher neurological metabolism, stronger protection against free radicals, and stronger neuroplasticity against neurological diseases. The resulting increase in brain function was due to the induced change in gene expression, increase in trophic factors, and reduction in pro-inflammatory cytokines.[53]
References
- Ebert SE, Jensen P, Ozenne B, Armand S, Svarer C, Stenbaek DS et al. Molecular imaging of neuroinflammation in patients after mild traumatic brain injury: a longitudinal 123 I-CLINDE SPECT study. Eur J Neurol 2019. doi:10.1111/ene.13971.
- Gendelman HE (December 2002). "Neural immunity: Friend or foe?". Journal of Neurovirology. 8 (6): 474–9. doi:10.1080/13550280290168631. PMID 12476342. S2CID 15631988.
- Das Sarma J (April 2014). "Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology". Journal of Neurovirology. 20 (2): 122–36. doi:10.1007/s13365-013-0188-4. PMID 23979705. S2CID 15223990.
- 't Hart BA, den Dunnen WF (September 2013). "Commentary on special issue: CNS diseases and the immune system". Journal of Neuroimmune Pharmacology. 8 (4): 757–9. doi:10.1007/s11481-013-9486-0. PMID 23754135.
- Streit WJ, Mrak RE, Griffin WS (July 2004). "Microglia and neuroinflammation: a pathological perspective". Journal of Neuroinflammation. 1 (1): 14. doi:10.1186/1742-2094-1-14. PMC 509427. PMID 15285801.
- Mayer CL, Huber BR, Peskind E (October 2013). "Traumatic brain injury, neuroinflammation, and post-traumatic headaches". Headache. 53 (9): 1523–30. doi:10.1111/head.12173. PMC 4089888. PMID 24090534.
- Tohidpour, Abolghasem; Morgun, Andrey V.; Boitsova, Elizaveta B.; Malinovskaya, Natalia A.; Martynova, Galina P.; Khilazheva, Elena D.; Kopylevich, Natalia V.; Gertsog, Galina E.; Salmina, Alla B. (2017-06-20). "Neuroinflammation and Infection: Molecular Mechanisms Associated with Dysfunction of Neurovascular Unit". Frontiers in Cellular and Infection Microbiology. 7: 276. doi:10.3389/fcimb.2017.00276. ISSN 2235-2988. PMC 5476750. PMID 28676848.
- Garden GA (October 2013). "Epigenetics and the modulation of neuroinflammation". Neurotherapeutics. 10 (4): 782–8. doi:10.1007/s13311-013-0207-4. PMC 3805872. PMID 23963788.
- Anderson MA, Burda JE, Sofroniew MV (April 2016). "Astrocyte scar formation aids central nervous system axon regeneration". Nature. 1 (1): 195–200. Bibcode:2016Natur.532..195A. doi:10.1038/nature17623. PMC 5243141. PMID 27027288.
- Ramesh G, MacLean AG, Philipp MT (July 2013). "Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain". Mediators of Inflammation. 2013: 480739. doi:10.1155/2013/480739. PMC 3753746. PMID 23997430.
- Ren H, Han R, Chen X, Liu X, Wan J, Wang L, Yang X, Wang J (May 2020). "Potential therapeutic targets for intracerebral hemorrhage-associated inflammation: An update". J Cereb Blood Flow Metab. 40 (9): 1752–1768. doi:10.1177/0271678X20923551. PMC 7446569. PMID 32423330.
- Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, Che F, Chen X, Ren H, Hong M, Wang J (March 2019). "Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage". Prog. Neurobiol. 178: 101610. doi:10.1016/j.pneurobio.2019.03.003. PMID 30923023. S2CID 85495400.
- DiSabato, Damon; Quan, Ning; Godbout, Jonathan P. (October 2016). "Neuroinflammation: The Devil is in the Details". Journal of Neurochemistry. 139 (Suppl 2): 136–153. doi:10.1111/jnc.13607. ISSN 0022-3042. PMC 5025335. PMID 26990767.
- Xiong, Ye; Mahmood, Asim; Chopp, Michael (June 2018). "Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities". Chinese Journal of Traumatology. 21 (3): 137–151. doi:10.1016/j.cjtee.2018.02.003. ISSN 1008-1275. PMC 6034172. PMID 29764704.
- Zhou X, He X, Ren Y (October 2014). "Function of microglia and macrophages in secondary damage after spinal cord injury". Neural Regeneration Research. 9 (20): 1787–95. doi:10.4103/1673-5374.143423. PMC 4239768. PMID 25422640.
- Garcia E, Aguilar-Cevallos J, Silva-Garcia R, Ibarra A (2016). "Cytokine and Growth Factor Activation In Vivo and In Vitro after Spinal Cord Injury". Mediators of Inflammation. 2016: 9476020. doi:10.1155/2016/9476020. PMC 4935915. PMID 27418745.
- Cameron MJ, Kelvin DJ (2013). Cytokines, Chemokines and Their Receptors. Landes Bioscience.
- Martinez FO, Gordon S (2014-03-03). "The M1 and M2 paradigm of macrophage activation: time for reassessment". F1000Prime Reports. 6 (13): 13. doi:10.12703/p6-13. PMC 3944738. PMID 24669294.
- Wang X, Cao K, Sun X, Chen Y, Duan Z, Sun L, Guo L, Bai P, Sun D, Fan J, He X, Young W, Ren Y (April 2015). "Macrophages in spinal cord injury: phenotypic and functional change from exposure to myelin debris". Glia. 63 (4): 635–51. doi:10.1002/glia.22774. PMC 4331228. PMID 25452166.
- Fehlings MG, Nguyen DH (May 2010). "Immunoglobulin G: a potential treatment to attenuate neuroinflammation following spinal cord injury". Journal of Clinical Immunology. 30 Suppl 1 (1): S109–12. doi:10.1007/s10875-010-9404-7. PMC 2883090. PMID 20437085.
- Gomes da Silva, Sergio (2013). "Exercise-induced hippocampal anti-inflammatory response in aged rats". Journal of Neuroinflammation. 10: 61. doi:10.1186/1742-2094-10-61. PMC 3657539. PMID 23663962.
- Janowitz D, Habes M, Toledo JB, Hannemann A, Frenzel S, Terock J, Davatzikos C, Hoffmann W, Grabe HJ (2019). "Inflammatory markers and imaging patterns of advanced brain aging in the general population". Brain Imaging and Behavior. 14 (4): 1108–1117. doi:10.1007/s11682-019-00058-y. PMC 8374834. PMID 30820858.
- Kohman RA, Bhattacharya TK, Wojcik E, Rhodes JS (September 2013). "Exercise reduces activation of microglia isolated from hippocampus and brain of aged mice". Journal of Neuroinflammation. 10: 114. doi:10.1186/1742-2094-10-114. PMC 3848770. PMID 24044641.
- Lynch MA (2010). "Age-related neuroinflammatory changes negatively impact on neuronal function". Frontiers in Aging Neuroscience. 1: 6. doi:10.3389/neuro.24.006.2009. PMC 2874409. PMID 20552057.
- Sparkman, Nathan L.; Johnson, Rodney W. (2008). "Neuroinflammation Associated with Aging Sensitizes the Brain to the Effects of Infection or Stress". Neuroimmunomodulation. 15 (4–6): 323–330. doi:10.1159/000156474. ISSN 1021-7401. PMC 2704383. PMID 19047808.
- Meraz-Ríos MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernández J, Campos-Peña V (August 2013). "Inflammatory process in Alzheimer's Disease". Frontiers in Integrative Neuroscience. 7: 59. doi:10.3389/fnint.2013.00059. PMC 3741576. PMID 23964211.
- Culibrk RA, Hahn MS (2020). "The Role of Chronic Inflammatory Bone and Joint Disorders in the Pathogenesis and Progression of Alzheimer's Disease". Frontiers in Aging Neuroscience. 12: 583884. doi:10.3389/fnagi.2020.583884. PMC 7750365. PMID 33364931.
- Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG (November 2009). "Does neuroinflammation fan the flame in neurodegenerative diseases?". Molecular Neurodegeneration. 4: 47. doi:10.1186/1750-1326-4-47. PMC 2784760. PMID 19917131.
- Imbimbo, Bruno P.; Solfrizzi, Vincenzo; Panza, Francesco (2010-05-21). "Are NSAIDs Useful to Treat Alzheimer's Disease or Mild Cognitive Impairment?". Frontiers in Aging Neuroscience. 2. doi:10.3389/fnagi.2010.00019. ISSN 1663-4365. PMC 2912027. PMID 20725517.
- Barnum CJ, Tansey MG (August 2012). "Neuroinflammation and non-motor symptoms: the dark passenger of Parkinson's disease?". Current Neurology and Neuroscience Reports. 12 (4): 350–8. doi:10.1007/s11910-012-0283-6. PMID 22580742. S2CID 46437442.
- Mejzini, Rita; Flynn, Loren L.; Pitout, Ianthe L.; Fletcher, Sue; Wilton, Steve D.; Akkari, P. Anthony (2019-12-06). "ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?". Frontiers in Neuroscience. 13: 1310. doi:10.3389/fnins.2019.01310. ISSN 1662-453X. PMC 6909825. PMID 31866818.
- Komine, Okiru; Yamanaka, Koji (November 2015). "Neuroinflammation in motor neuron disease". Nagoya Journal of Medical Science. 77 (4): 537–549. ISSN 0027-7622. PMC 4664586. PMID 26663933.
- Turner, M.R; Cagnin, A; Turkheimer, F.E; Miller, C.C.J; Shaw, C.E; Brooks, D.J; Leigh, P.N; Banati, R.B (April 2004). "Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study". Neurobiology of Disease. 15 (3): 601–609. doi:10.1016/j.nbd.2003.12.012. PMID 15056468. S2CID 32760657.
- Pun, Frank W.; Liu, Bonnie Hei Man; Long, Xi; Leung, Hoi Wing; Leung, Geoffrey Ho Duen; Mewborne, Quinlan T.; Gao, Junli; Shneyderman, Anastasia; Ozerov, Ivan V.; Wang, Ju; Ren, Feng (2022-06-28). "Identification of Therapeutic Targets for Amyotrophic Lateral Sclerosis Using PandaOmics – An AI-Enabled Biological Target Discovery Platform". Frontiers in Aging Neuroscience. 14: 914017. doi:10.3389/fnagi.2022.914017. ISSN 1663-4365. PMC 9273868. PMID 35837482.
- Gargiulo, S.; Anzilotti, S.; Coda, A. R. D.; Gramanzini, M.; Greco, A.; Panico, M.; Vinciguerra, A.; Zannetti, A.; Vicidomini, C.; Dollé, F.; Pignataro, G. (July 2016). "Imaging of brain TSPO expression in a mouse model of amyotrophic lateral sclerosis with 18F-DPA-714 and micro-PET/CT". European Journal of Nuclear Medicine and Molecular Imaging. 43 (7): 1348–1359. doi:10.1007/s00259-016-3311-y. ISSN 1619-7070. PMID 26816193. S2CID 12200161.
- Geloso, Maria Concetta; Corvino, Valentina; Marchese, Elisa; Serrano, Alessia; Michetti, Fabrizio; D’Ambrosi, Nadia (2017-07-25). "The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches". Frontiers in Aging Neuroscience. 9: 242. doi:10.3389/fnagi.2017.00242. ISSN 1663-4365. PMC 5524666. PMID 28790913.
- Izrael, Michal; Slutsky, Shalom Guy; Revel, Michel (2020-07-28). "Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease". Frontiers in Neuroscience. 14: 824. doi:10.3389/fnins.2020.00824. ISSN 1662-453X. PMC 7399224. PMID 32848579.
- Beers, David R.; Henkel, Jenny S.; Xiao, Qin; Zhao, Weihua; Wang, Jinghong; Yen, Albert A.; Siklos, Laszlo; McKercher, Scott R.; Appel, Stanley H. (2006-10-24). "Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis". Proceedings of the National Academy of Sciences. 103 (43): 16021–16026. Bibcode:2006PNAS..10316021B. doi:10.1073/pnas.0607423103. ISSN 0027-8424. PMC 1613228. PMID 17043238.
- Lepore, Angelo C; Rauck, Britta; Dejea, Christine; Pardo, Andrea C; Rao, Mahendra S; Rothstein, Jeffrey D; Maragakis, Nicholas J (November 2008). "Focal transplantation–based astrocyte replacement is neuroprotective in a model of motor neuron disease". Nature Neuroscience. 11 (11): 1294–1301. doi:10.1038/nn.2210. ISSN 1097-6256. PMC 2656686. PMID 18931666.
- Hooten, Kristopher G.; Beers, David R.; Zhao, Weihua; Appel, Stanley H. (April 2015). "Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis". Neurotherapeutics. 12 (2): 364–375. doi:10.1007/s13311-014-0329-3. ISSN 1933-7213. PMC 4404435. PMID 25567201.
- Bowerman, Melissa; Vincent, Thierry; Scamps, Frédérique; Perrin, Florence E.; Camu, William; Raoul, Cédric (2013). "Neuroimmunity dynamics and the development of therapeutic strategies for amyotrophic lateral sclerosis". Frontiers in Cellular Neuroscience. 7: 214. doi:10.3389/fncel.2013.00214. ISSN 1662-5102. PMC 3833095. PMID 24312006.
- Beers, David R.; Henkel, Jenny S.; Zhao, Weihua; Wang, Jinghong; Appel, Stanley H. (2008-10-07). "CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS". Proceedings of the National Academy of Sciences. 105 (40): 15558–15563. Bibcode:2008PNAS..10515558B. doi:10.1073/pnas.0807419105. ISSN 0027-8424. PMC 2547419. PMID 18809917.
- Henkel, Jenny S.; Beers, David R.; Wen, Shixiang; Rivera, Andreana L.; Toennis, Karen M.; Appel, Joan E.; Zhao, Weihua; Moore, Dan H.; Powell, Suzanne Z.; Appel, Stanley H. (January 2013). "Regulatory T‐lymphocytes mediate amyotrophic lateral sclerosis progression and survival". EMBO Molecular Medicine. 5 (1): 64–79. doi:10.1002/emmm.201201544. ISSN 1757-4676. PMC 3569654. PMID 23143995.
- Mantovani, Stefania; Garbelli, Silvia; Pasini, Alessandra; Alimonti, Dario; Perotti, Cesare; Melazzini, Mario; Bendotti, Caterina; Mora, Gabriele (May 2009). "Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process". Journal of Neuroimmunology. 210 (1–2): 73–79. doi:10.1016/j.jneuroim.2009.02.012. PMID 19307024. S2CID 11379824.
- Zondler, Lisa; Müller, Kathrin; Khalaji, Samira; Bliederhäuser, Corinna; Ruf, Wolfgang P.; Grozdanov, Veselin; Thiemann, Meinolf; Fundel-Clemes, Katrin; Freischmidt, Axel; Holzmann, Karlheinz; Strobel, Benjamin (September 2016). "Peripheral monocytes are functionally altered and invade the CNS in ALS patients". Acta Neuropathologica. 132 (3): 391–411. doi:10.1007/s00401-016-1548-y. ISSN 1432-0533. PMID 26910103. S2CID 23120645.
- Butovsky, Oleg; Siddiqui, Shafiuddin; Gabriely, Galina; Lanser, Amanda J.; Dake, Ben; Murugaiyan, Gopal; Doykan, Camille E.; Wu, Pauline M.; Gali, Reddy R.; Iyer, Lakshmanan K.; Lawson, Robert (2012-09-04). "Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS". Journal of Clinical Investigation. 122 (9): 3063–3087. doi:10.1172/JCI62636. ISSN 0021-9738. PMC 3428086. PMID 22863620.
- Mantovani, S.; Gordon, R.; Macmaw, J.K.; Pfluger, C.M.M.; Henderson, R.D.; Noakes, P.G.; McCombe, P.A.; Woodruff, T.M. (November 2014). "Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood". Journal of Neuroimmunology. 276 (1–2): 213–218. doi:10.1016/j.jneuroim.2014.09.005. PMID 25262158. S2CID 24553591.
- Heurich, Bianca; el Idrissi, Nawal Bahia; Donev, Rossen M.; Petri, Susanne; Claus, Peter; Neal, James; Morgan, B. Paul; Ramaglia, Valeria (June 2011). "Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis". Journal of Neuroimmunology. 235 (1–2): 104–109. doi:10.1016/j.jneuroim.2011.03.011. PMID 21501881. S2CID 30757392.
- "Multiple Sclerosis: Hope Through Research". The National Institute of Neurological Disorders and Stroke. Retrieved 2016-08-22.
- Zindler E, Zipp F (December 2010). "Neuronal injury in chronic CNS inflammation". Best Practice & Research. Clinical Anaesthesiology. 24 (4): 551–62. doi:10.1016/j.bpa.2010.11.001. PMID 21619866.
- McPherson RC, Anderton SM (September 2013). "Adaptive immune responses in CNS autoimmune disease: mechanisms and therapeutic opportunities". Journal of Neuroimmune Pharmacology. 8 (4): 774–90. doi:10.1007/s11481-013-9453-9. PMID 23568718. S2CID 16198820.
- Seo, Dae-Yun; Heo, Jun-Won; Ko, Jeong Rim; Kwak, Hyo-Bum (November 2019). "Exercise and Neuroinflammation in Health and Disease". International Neurourology Journal. 23 (Suppl 2): S82–92. doi:10.5213/inj.1938214.107. ISSN 2093-4777. PMC 6905205. PMID 31795607.
- Seo, Dae-Yun; Heo, Jun-Won; Ko, Jeong Rim; Kwak, Hyo-Bum (November 2019). "Exercise and Neuroinflammation in Health and Disease". International Neurourology Journal. 23 (Suppl 2): S82–92. doi:10.5213/inj.1938214.107. ISSN 2093-4777. PMC 6905205. PMID 31795607.
Further reading
- Maggi P, Macri SM, Gaitán MI, Leibovitch E, Wholer JE, Knight HL, Ellis M, Wu T, Silva AC, Massacesi L, Jacobson S, Westmoreland S, Reich DS (October 2014). "The formation of inflammatory demyelinated lesions in cerebral white matter". Annals of Neurology. 76 (4): 594–608. doi:10.1002/ana.24242. PMC 4723108. PMID 25088017.