Pyroptosis
Pyroptosis is a highly inflammatory form of lytic programmed cell death that occurs most frequently upon infection with intracellular pathogens and is likely to form part of the antimicrobial response. This process promotes the rapid clearance of various bacterial, viral, fungal and protozoan infections by removing intracellular replication niches and enhancing the host's defensive responses. Pyroptosis can take place in immune cells and is also reported to occur in keratinocytes and some epithelial cells.[1]
The process is initiated by formation of a large supramolecular complex termed the inflammasome (also known as a pyroptosome) upon intracellular danger signals.[2] The inflammasome activates a different set of caspases as compared to apoptosis, for example, caspase-1/4/5 in humans and caspase-11 in mice.[3] These caspases contribute to the maturation and activation of several proinflammatory cytokines and pore-forming protein gasdermins. Formation of pores causes cell membrane rupture and release of cytokines, as well as various damage-associated molecular pattern (DAMP) molecules such as HMGB-1, ATP and DNA, out of the cell. These molecules recruit more immune cells and further perpetuate the inflammatory cascade in the tissue.[4][5]
However, in pathogenic chronic diseases, the inflammatory response does not eradicate the primary stimulus. A chronic form of inflammation ensues that ultimately contributes to tissue damage. Pyroptosis is associated with diseases including cancer, neurodegeneration and those of the cardiovascular system. Some examples of pyroptosis include Salmonella-infected macrophages and abortively HIV-infected T helper cells.[6][7][8]
Discovery
This type of inherently pro-inflammatory programmed cell death was named pyroptosis in 2001 by Dr. Brad T. Cookson, an associate professor of microbiology and laboratory medicine at the University of Washington.[9] The Greek pyro refers to fire and ptosis means falling. The compound term of pyroptosis may be understood as "fiery falling", which describes the bursting of pro-inflammatory chemical signals from the dying cell. Pyroptosis has a distinct morphology and mechanism compared to those of other forms of cell death.[10] It has been suggested that microbial infection was the main evolutionary pressure for this pathway.[11] In 2013, caspase-11 dependent noncanonical pathway was discovered, suggesting lipopolysaccharides (LPS) can trigger pyroptosis and subsequent inflammatory responses independent of toll-like receptor 4 (TLR4).[12] In 2015, gasdermin D (GSDMD) was identified as the effector of pyroptosis that permeabilizes the cell membrane.[3][13] In 2021, the high-resolution structure of the GSDMD pore was solved by cryo-electron microscopy (cryo-EM).[14]
Morphological characteristics
Pyroptosis, as a form of programmed cell death, has many morphological differences as compared to apoptosis. Both pyroptosis and apoptosis undergo chromatin condensation, but during apoptosis, the nucleus breaks into multiple chromatin bodies; in pyroptosis, the nucleus remains intact.[15] In a cell that undergoes pyroptosis, gasdermin pores are formed on the plasma membrane, resulting in water influx and cell lysis.[1][16]
In terms of mechanism, pyroptosis is activated by inflammatory caspases, including caspase-1/4/5 in humans and caspase-11 in mice. Pro-apoptotic caspases, including caspase-6/7/8/9, are not required for pyroptosis. Caspase-3 activation can take place in both apoptosis and pyroptosis.[1][16]
Although both pyroptosis and necroptosis are triggered by membrane pore formation, pyroptosis is more controlled. Cells that undergo pyroptosis exhibit membrane blebbing and produce protrusions known as pyroptotic bodies, a process not found in necroptosis.[17] Also, necroptosis works in a caspase-independent fashion. It is proposed that both pyroptosis and necroptosis may act as defence systems against pathogens when apoptotic pathways are blocked.
| Characteristics | Apoptosis | Pyroptosis | Necroptosis | |
|---|---|---|---|---|
| Morphology | Cell lysis | NO | YES | YES |
| Cell swelling | NO | YES | YES | |
| Pore formation | NO | YES | YES | |
| Membrane blebbing | YES | YES | NO | |
| DNA fragmentation | YES | YES | YES | |
| Nucleus intact | NO | YES | NO | |
| Mechanism | Caspase-1 activation | NO | YES | NO |
| Caspase-3 activation | YES | YES | NO | |
| GSDMD activation | NO | YES | NO | |
| Outcome | Inflammation | NO | YES | YES |
| Programmed cell death | YES | YES | YES |
Mechanism
The innate immune system, by using germ-line encoded pattern recognition receptors (PRRs), can recognize a wide range of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) upon microbe infection. Classic examples of PRRs include toll-like receptors (TLRs) and NOD-like receptors (NLRs).[18] Recognition of PAMPs and DAMPs triggers the formation of multi-protein complex inflammasomes, which then activates caspases to initiate pyroptosis. The inflammasome pathway may be canonical or noncanonical, with the former using caspase-1-activating inflammasomes and the latter using other caspases.[19]
The canonical inflammasome pathway
In the canonical inflammasome pathway, PAMPs and DAMPs are recognised by certain endogenous PRRs. For example, NLR proteins NLRC4 can recognise flagellin and type III secretion system components.[20] NLRP3 is activated by cellular events induced by different PAMPs and DAMPs stimuli.[21] Some non-NLR proteins like absent in melanoma 2 (AIM2) and pyrin can also be activated and form inflammasomes.[19] Also, non-inflammasome-forming PRRs such as TLRs, NOD1 and NOD2 also play important roles in pyroptosis. These receptors upregulate expression of inflammatory cytokines such as IFN α/β, tumour necrosis factor (TNF), IL-6 and IL-12 through NF-κB and MAPK-signaling pathways. In addition, pro-IL-1β and pro-IL-18 is released to be processed by cysteine-mediated caspase-1.[22][23]
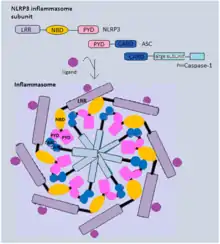
Canonical inflammasomes mostly contain three components: a sensor protein (PRRs), an adaptor (ASC) and an effector (caspase-1).[19] Generally, inflammasome-forming NLR proteins share a similar structure, several leucine-rich repeat (LRR) domains, a central nucleotide-binding and oligomerization domain (NBD) and an N-terminal pyrin domain (PYD). NLRP3, for example, recruits ASC adaptor protein via PYD-PYD interaction. Both pro-caspase-1 and ASC contain a caspase activation and recruitment domain (CARD), and this homotypic CARD-CARD interaction enables autocatalytic cleavage and reassembly of procaspase-1 to form active caspase-1.[24] Alternatively, NLRC4 can directly recruit pro-caspase-1, as it has a CARD domain instead of a pyrin domain.[25]
Activated caspase-1 is responsible for cleavage of pro-IL-1β and pro-IL-18. These cytokines, once processed, will be in their biologically active form ready to be released from the host cells. In addition, caspase-1 also cleaves the cytosolic gasdermin D (GSDMD). GSDMD can be cleaved to produce an N-terminal domain (GSDMD-N) and a C-terminal domain (GSDMD-C). GSDMD-N can oligomerize and form transmembrane pores that have an inner diameter of 10-14 nm.[26] The pores allow secretion of IL-1β and IL-18 and various cytosolic content to extracellular space, and they also disrupt the cellular ionic gradient. The resulting increase in osmotic pressure causes an influx of water followed by cell swelling and bursting. Notably, GSDMD-N is autoinhibited by GSDMD C-terminal domain before cleavage to prevent cell lysis in normal conditions.[27] Also, GSDMD-N can only insert itself into the inner membrane with specific lipid compositions,[28] which limits its damage to neighbour cells.
The noncanonical inflammasome pathway
The noncanonical inflammasome pathway is initiated by binding of lipopolysaccharide (LPS) of gram-negative bacteria directly onto caspase-4/5 in humans and caspase-11 in murines. Binding of LPS onto these caspases promotes their oligomerization and activation.[12] These caspases can cleave GSDMD to release GSDMD-N and trigger pyroptosis. In addition, an influx of potassium ions upon membrane permeabilization triggers activation of NLRP3, which then leads to formation of NLRP3 inflammasome and activation of caspase-1.[19] These processes facilitate the cleavage of GSDMD and promote the maturation and release of pro-inflammatory cytokines.
Caspase-3-dependent pyroptotic pathway
An alternative pathway that links apoptosis and pyroptosis has been recently proposed. Caspase-3, an executioner caspase in apoptosis, can cleave gasdermin E (GSDME) to produce a N-terminal fragment and a C-terminal fragment in a way similar to GSDMD cleavage.[3] When apoptotic cells are not scavenged by macrophages, GSDME expression is then upregulated by p53. GSDME is then activated by caspase-3 to form pores on the cell membrane. It has also been found that GSDME can permeabilise mitochondrial membranes to release cytochrome c, which further activates caspase-3 and accelerates GSDME cleavage.[29] This positive feedback loop ensures that programmed cell death is carried forward.
Clinical relevance
Pyroptosis acts as a defence mechanism against infection by inducing pathological inflammation. The formation of inflammasomes and the activity of caspase-1 determine the balance between pathogen resolution and disease.
In a healthy cell, caspase-1 activation helps to fight infection caused by Salmonella and Shigella by introducing cell death to restrict pathogen growth.[6] When the "danger" signal is sensed, the quiescent cells will be activated to undergo pyroptosis and produce inflammatory cytokines IL-1β and IL-18. IL-18 will stimulate IFNγ production and initiates the development of TH1 responses. (TH1 responses tend to release cytokines that direct an immediate removal of the pathogen.)[30] The cell activation results in an increase in cytokine levels, which will augment the consequences of inflammation and this, in turn, contributes to the development of the adaptive response as infection progresses. The ultimate resolution will clear pathogens.
In contrast, persistent inflammation will produce excessive immune cells, which is detrimental. If the amplification cycles persist, metabolic disorder, autoinflammatory diseases and liver injury associated with chronic inflammation will occur.[30]
Cancer
Pyroptosis, as an inflammation-associated programmed cell death, has wide implications in various cancer types. Principally, pyroptosis can kill cancer cells and inhibit tumour development in the presence of endogenous DAMPs. In some cases, GSDMD can be used as a prognostic marker for cancers. However, prolonged production of inflammatory bodies may facilitate the formation of microenvironments that favour tumour growth.[31] Understanding the mechanisms of pyroptosis and identifying pyroptosis-associated molecules can be useful in treating different cancers.
In gastric cancer cells, presence of GSDMD can inhibit cyclin A2/CDK2 complexes, leading to cell cycle arrest and thus inhibit tumour development. Also, cellular concentration of GSDME increases when gastric cancer cells are treated with certain chemotherapy drugs. GSDME then activates caspase-3 and triggers pyroptotic cell death.[16]
Cervical cancer can be caused by human papillomavirus (HPV) infection. AIM2 protein can recognise viral DNA in cytoplasm and form AIM2 inflammasome, which then triggers by a caspase-1 dependent canonical pyroptosis pathway. HPV infection causes the upregulation of sirtuin 1 protein, which disrupts the transcription factor for AIM2, RelB. Knockdown of sirtuin 1 upregulates AIM2 expression and triggers pyroptosis.[32]
Metabolic disorder
The level of expression of NLRP3 inflammasome and caspase-1 has a direct relation to the severity of several metabolic syndromes, such as obesity and type II diabetic mellitus (T2DM). This is because the subsequent production level of IL-1β and IL-18, cytokines that impair the secretion of insulin, is affected by the activity of caspase-1. Glucose uptake level is then diminished, and the condition is known as insulin resistance.[33] The condition is further accelerated by the IL-1β-induced destruction of pancreatic β cells.[34]
Cryopyrinopathies
A mutation in the gene coding of inflammasomes leads to a group of autoinflammatory diseases called cryopyrinopathies. This group includes Muckle–Wells syndrome, cold autoinflammatory syndrome and chronic infantile neurologic cutaneous and articular syndrome, all showing symptoms of sudden fevers and localized inflammation.[35] The mutated gene in such cases is the NLRP3, impeding the activation of inflammasome and resulting in an excessive production of IL-1β. This effect is known as "gain-of-function".[36]
HIV and AIDS
Recent studies demonstrate that caspase-1-mediated pyroptosis drives CD4 T-cell depletion and inflammation by HIV,[7][37][38][39] two signature events that propel HIV disease progression to AIDS. Although pyroptosis contributes to the host's ability to rapidly limit and clear infection by removing intracellular replication niches and enhancing defensive responses through the release of proinflammatory cytokines and endogenous danger signals, in pathogenic inflammation, such as that elicited by HIV-1, this beneficial response does not eradicate the primary stimulus. In fact, it appears to create a pathogenic vicious cycle in which dying CD4 T cells release inflammatory signals that attract more cells into the infected lymphoid tissues to die and to produce chronic inflammation and tissue injury. It may be possible to break this pathogenic cycle with safe and effective caspase-1 inhibitors. These agents could form a new and exciting ‘anti-AIDS' therapy for HIV-infected subjects in which the treatment targets the host instead of the virus. Of note, Caspase-1 deficient mice develop normally,[40][41] arguing that inhibition of this protein would produce beneficial rather than harmful therapeutic effects in HIV patients. Recently, pyroptosis and downstream pathways were identified as promising targets for treatment of severe COVID-19-associated diseases.[42]
References
- Jorgensen I, Miao EA (May 2015). "Pyroptotic cell death defends against intracellular pathogens". Immunological Reviews. 265 (1): 130–42. doi:10.1111/imr.12287. PMC 4400865. PMID 25879289.
- Nirmala JG, Lopus M (April 2020). "Cell death mechanisms in eukaryotes". Cell Biology and Toxicology. 36 (2): 145–164. doi:10.1007/s10565-019-09496-2. PMID 31820165. S2CID 208869679.
- Gong W, Shi Y, Ren J (March 2020). "Research progresses of molecular mechanism of pyroptosis and its related diseases". Immunobiology. 225 (2): 151884. doi:10.1016/j.imbio.2019.11.019. PMID 31822435. S2CID 209314359.
- Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. (August 2014). "The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response". Nature Immunology. 15 (8): 738–48. doi:10.1038/ni.2919. PMID 24952504. S2CID 6928042.
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. (August 2014). "The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation". Nature Immunology. 15 (8): 727–37. doi:10.1038/ni.2913. PMC 4116676. PMID 24952505.
- Fink SL, Cookson BT (November 2006). "Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages". Cellular Microbiology. 8 (11): 1812–25. doi:10.1111/j.1462-5822.2006.00751.x. PMID 16824040.
- Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. (January 2014). "Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection". Nature. 505 (7484): 509–14. Bibcode:2014Natur.505..509D. doi:10.1038/nature12940. PMC 4047036. PMID 24356306.
- Doitsh G, Greene WC (March 2016). "Dissecting How CD4 T Cells Are Lost During HIV Infection". Cell Host & Microbe. 19 (3): 280–91. doi:10.1016/j.chom.2016.02.012. PMC 4835240. PMID 26962940.
- Cookson BT, Brennan MA (March 2001). "Pro-inflammatory programmed cell death". Trends in Microbiology. 9 (3): 113–4. doi:10.1016/S0966-842X(00)01936-3. PMID 11303500.
- Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P (November 2009). "Major cell death pathways at a glance". Microbes and Infection. 11 (13): 1050–62. doi:10.1016/j.micinf.2009.08.013. PMID 19733681.
- Dagenais M, Skeldon A, Saleh M (January 2012). "The inflammasome: in memory of Dr. Jurg Tschopp". Cell Death and Differentiation. 19 (1): 5–12. doi:10.1038/cdd.2011.159. PMC 3252823. PMID 22075986.
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. (October 2014). "Inflammatory caspases are innate immune receptors for intracellular LPS". Nature. 514 (7521): 187–92. Bibcode:2014Natur.514..187S. doi:10.1038/nature13683. PMID 25119034. S2CID 4459091.
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. (October 2015). "Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death". Nature. 526 (7575): 660–5. Bibcode:2015Natur.526..660S. doi:10.1038/nature15514. PMID 26375003. S2CID 4407455.
- Xia, Shiyu; Zhang, Zhibin; Magupalli, Venkat Giri; Pablo, Juan Lorenzo; Dong, Ying; Vora, Setu M.; Wang, Longfei; Fu, Tian-Min; Jacobson, Matthew P.; Greka, Anna; Lieberman, Judy (2021-04-21). "Gasdermin D pore structure reveals preferential release of mature interleukin-1". Nature. 593 (7860): 607–611. Bibcode:2021Natur.593..607X. doi:10.1038/s41586-021-03478-3. ISSN 0028-0836. PMC 8588876. PMID 33883744.
- Brennan MA, Cookson BT (October 2000). "Salmonella induces macrophage death by caspase-1-dependent necrosis". Molecular Microbiology. 38 (1): 31–40. doi:10.1046/j.1365-2958.2000.02103.x. PMID 11029688. S2CID 30022137.
- Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. (January 2020). "Pyroptosis: A new frontier in cancer". Biomedicine & Pharmacotherapy. 121: 109595. doi:10.1016/j.biopha.2019.109595. PMID 31710896.
- Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. (September 2016). "Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis". Cell Research. 26 (9): 1007–20. doi:10.1038/cr.2016.100. PMC 5034106. PMID 27573174.
- Bortoluci KR, Medzhitov R (May 2010). "Control of infection by pyroptosis and autophagy: role of TLR and NLR". Cellular and Molecular Life Sciences. 67 (10): 1643–51. doi:10.1007/s00018-010-0335-5. PMID 20229126. S2CID 20682184.
- Platnich JM, Muruve DA (July 2019). "NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways". Archives of Biochemistry and Biophysics. 670: 4–14. doi:10.1016/j.abb.2019.02.008. PMID 30772258. S2CID 73464235.
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. (September 2011). "The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus". Nature. 477 (7366): 596–600. Bibcode:2011Natur.477..596Z. doi:10.1038/nature10510. PMID 21918512. S2CID 4429240.
- Kelley N, Jeltema D, Duan Y, He Y (July 2019). "The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation". International Journal of Molecular Sciences. 20 (13): 3328. doi:10.3390/ijms20133328. PMC 6651423. PMID 31284572.
- Kawai T, Akira S (May 2006). "TLR signaling". Cell Death and Differentiation. 13 (5): 816–25. doi:10.1038/sj.cdd.4401850. PMID 16410796.
- Mukherjee T, Hovingh ES, Foerster EG, Abdel-Nour M, Philpott DJ, Girardin SE (July 2019). "NOD1 and NOD2 in inflammation, immunity and disease". Archives of Biochemistry and Biophysics. 670: 69–81. doi:10.1016/j.abb.2018.12.022. PMID 30578751. S2CID 58621835.
- Broz P, Dixit VM (July 2016). "Inflammasomes: mechanism of assembly, regulation and signalling". Nature Reviews. Immunology. 16 (7): 407–20. doi:10.1038/nri.2016.58. PMID 27291964. S2CID 32414010.
- Duncan JA, Canna SW (January 2018). "The NLRC4 Inflammasome". Immunological Reviews. 281 (1): 115–123. doi:10.1111/imr.12607. PMC 5897049. PMID 29247997.
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. (July 2016). "Pore-forming activity and structural autoinhibition of the gasdermin family". Nature. 535 (7610): 111–6. Bibcode:2016Natur.535..111D. doi:10.1038/nature18590. PMID 27281216. S2CID 4391444.
- Liu Z, Wang C, Rathkey JK, Yang J, Dubyak GR, Abbott DW, Xiao TS (May 2018). "Structures of the Gasdermin D C-Terminal Domains Reveal Mechanisms of Autoinhibition". Structure. 26 (5): 778–784.e3. doi:10.1016/j.str.2018.03.002. PMC 5932255. PMID 29576317.
- Qiu S, Liu J, Xing F (April 2017). "'Hints' in the killer protein gasdermin D: unveiling the secrets of gasdermins driving cell death". Cell Death and Differentiation. 24 (4): 588–596. doi:10.1038/cdd.2017.24. PMC 5384029. PMID 28362726.
- Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES (April 2019). "Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation". Nature Communications. 10 (1): 1689. Bibcode:2019NatCo..10.1689R. doi:10.1038/s41467-019-09397-2. PMC 6459836. PMID 30976076.
- Davis BK, Wen H, Ting JP (2011). "The inflammasome NLRs in immunity, inflammation, and associated diseases". Annual Review of Immunology. 29 (1): 707–35. doi:10.1146/annurev-immunol-031210-101405. PMC 4067317. PMID 21219188.
- Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, Hu J (September 2019). "The role of pyroptosis in cancer: pro-cancer or pro-"host"?". Cell Death & Disease. 10 (9): 650. doi:10.1038/s41419-019-1883-8. PMC 6733901. PMID 31501419.
- So D, Shin HW, Kim J, Lee M, Myeong J, Chun YS, Park JW (September 2018). "Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense". Oncogene. 37 (38): 5191–5204. doi:10.1038/s41388-018-0339-4. PMID 29844574. S2CID 44064866.
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. (February 2011). "The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance". Nature Medicine. 17 (2): 179–88. doi:10.1038/nm.2279. PMC 3076025. PMID 21217695.
- Strowig T, Henao-Mejia J, Elinav E, Flavell R (January 2012). "Inflammasomes in health and disease". Nature. 481 (7381): 278–86. Bibcode:2012Natur.481..278S. doi:10.1038/nature10759. PMID 22258606. S2CID 205227460.
- Neven B, Prieur AM, Quartier dit Maire P (September 2008). "Cryopyrinopathies: update on pathogenesis and treatment". Nature Clinical Practice. Rheumatology. 4 (9): 481–9. doi:10.1038/ncprheum0874. PMID 18665151. S2CID 13022253.
- Church LD, Cook GP, McDermott MF (January 2008). "Primer: inflammasomes and interleukin 1beta in inflammatory disorders". Nature Clinical Practice. Rheumatology. 4 (1): 34–42. doi:10.1038/ncprheum0681. PMID 18172447. S2CID 19986204.
- Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC (January 2014). "IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV". Science. 343 (6169): 428–32. Bibcode:2014Sci...343..428M. doi:10.1126/science.1243640. PMC 3976200. PMID 24356113.
- Lu, Wuxun; Demers, Andrew J.; Ma, Fangrui; Kang, Guobin; Yuan, Zhe; Wan, Yanmin; Li, Yue; Xu, Jianqing; Lewis, Mark; Li, Qingsheng (2016-01-15). Silvestri, G. (ed.). "Next-Generation mRNA Sequencing Reveals Pyroptosis-Induced CD4 + T Cell Death in Early Simian Immunodeficiency Virus-Infected Lymphoid Tissues". Journal of Virology. 90 (2): 1080–1087. doi:10.1128/JVI.02297-15. ISSN 0022-538X. PMC 4702687. PMID 26559826.
- Zhang, Chao; Song, Jin-Wen; Huang, Hui-Huang; Fan, Xing; Huang, Lei; Deng, Jian-Ning; Tu, Bo; Wang, Kun; Li, Jing; Zhou, Ming-Ju; Yang, Cui-Xian (2021-03-15). "NLRP3 inflammasome induces CD4+ T cell loss in chronically HIV-1–infected patients". The Journal of Clinical Investigation. 131 (6). doi:10.1172/JCI138861. ISSN 0021-9738. PMC 7954596. PMID 33720048.
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA (March 1995). "Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme". Science. 267 (5206): 2000–3. Bibcode:1995Sci...267.2000K. doi:10.1126/science.7535475. PMID 7535475.
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. (February 1995). "Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock". Cell. 80 (3): 401–11. doi:10.1016/0092-8674(95)90490-5. PMID 7859282. S2CID 18756273.
- Yap, Jeremy K. Y.; Moriyama, Miyu; Iwasaki, Akiko (2020-07-15). "Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19". Journal of Immunology. 205 (2): 307–312. doi:10.4049/jimmunol.2000513. ISSN 1550-6606. PMC 7343621. PMID 32493814.