Quality control in tissue engineering
The rapid development in the multidisciplinary field of tissue engineering has resulted in a variety of new and innovative medicinal products, often carrying living cells, intended to repair, regenerate or replace damaged human tissue. Tissue engineered medicinal products (TEMPs) vary in terms of the type and origin of cells and the product’s complexity. As all medicinal products, the safety and efficacy of TEMPs must be consistent throughout the manufacturing process. Quality control and assurance are of paramount importance and products are constantly assessed throughout the manufacturing process to ensure their safety, efficacy, consistency and reproducibility between batches.[1] The European Medicines Agency (EMA) is responsible for the development, assessment and supervision of medicines in the EU. The appointed committees are involved in referral procedures concerning safety or the balance of benefit/risk of a medicinal product. In addition, the committees organize inspections with regards to the conditions under which medicinal products are being manufactured. For example, the compliance with good manufacturing practice (GMP), good clinical practice (GCP), good laboratory practice (GLP) and pharmacovigilance (PhV).[2]
Risk assessment
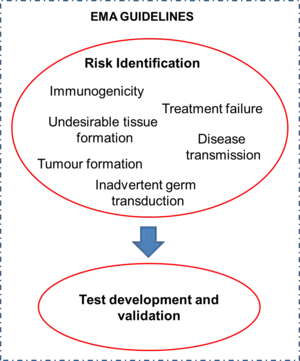
When quality control of TEMPs is considered, a risk assessment needs to be conducted. A risk is defined as a "potentially unfavourable effect that can be attributed to the clinical use of advanced therapy medicinal products (ATMPs) and is of concern to the patient and/or to other populations (e.g. caregivers and off-spring)".[3] Some risks include immunogenicity, disease transmission, tumor formation, treatment failure, undesirable tissue formation, and inadvertent germ transduction. A risk factor is defined as a "qualitative or quantitative characteristic that contributes to a specific risk following handling and/or administration of an ATMP".[3] The integration of all available information on risks and risk factors is called risk profiling. Due to the fact that every TEMP is different, the risks associated with each one of them vary and, subsequently, the procedures that must be implemented to ensure its quality are also unique to the product. Once the risks associated with the TEMP are identified, the appropriate tests must be developed and validated accordingly. Thus, there is no standard set of tests for the quality control of TEMPs. The EMA has released a set of regulatory guidelines on the topics to be considered by companies involved in the development and marketing of medicines for use in the European Union.[3][4][5][6] These guidelines have to be followed in order for the marketing authorization of a product to be issued. Fictitious examples of risk analysis for further elucidation of the process are provided in the EMA guidelines.[3]
Quality considerations
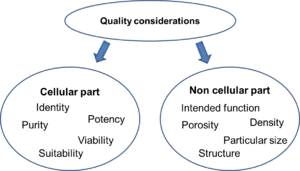
Careful and detailed documentation concerning the characteristics of the starting materials (e.g. history of the cell line derivation and cell banking) and manufacturing process steps (e.g. procurement of tissue or cells and manipulation) must be maintained. The cellular part of every cell-based medicinal product must be characterized in terms of identity, purity, potency, viability and suitability for the intended use. The non-cellular constituents must be also characterized with regards to their intended function in the final product. For example, scaffolds or membranes that are used to support the cells must be identified and characterized in terms of porosity, density, microscopic structure and particular size. The same requirement for characterization applies for biologically active molecules, such as growth factors or cytokines.[5]
Release specifications
Proper quality control involves the release testing of the final product through updated and validated methods. The release specifications of the product must be selected on the basis of the parameters defined during the characterization studies and the appropriate release tests must be performed. In case a release test cannot be performed on the final product but only on previous stages of the manufacturing, exceptions can be made after proper justification. However, in these cases adequate quality control has to rise from the manufacturing process. Specifications about the stability of the product, the presence or not of genetically modified cells, structural components and whether it is a combination product must also be defined.[5]
References
- Standardization In Cell And Tissue Engineering: Methods And Protocols Woodhead Publishing Series In Biomaterials, Edited By Vedih Salih, Elsevier 2013
- "European Medicines Agency - About Us - What we do". www.ema.europa.eu. Retrieved 2016-01-05.
- Guideline On The Risk-Based Approach According To Annex I, Part IV Of Directive 2001/83/EC For ATMPs, Committee For Advanced Therapies (CAT), 11 February 2013
- Guideline On Human Cell-Based Medicinal Products, Committee For Medicinal Product For Human Use (CHMP), London, 21 May 2008
- Reflection Paper On Stem Cell-Based Medicinal Products, Committee For Advanced Therapies (CAT), 14 January 2011
- Reflection Paper On Clinical Aspects Related To Tissue Engineered Products, Committee For Advanced Therapies (CAT), 19 March 2012