The Hallmarks of Cancer
The hallmarks of cancer were originally six biological capabilities acquired during the multistep development of human tumors and have since been increased to eight capabilities and two enabling capabilities. The idea was coined by Douglas Hanahan and Robert Weinberg in their paper "The Hallmarks of Cancer" published January 2000 in Cell.[1]
These hallmarks constitute an organizing principle for rationalizing the complexities of neoplastic disease. They include sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis. Underlying these hallmarks are genome instability, which generates the genetic diversity that expedites their acquisition, and inflammation, which fosters multiple hallmark functions. In addition to cancer cells, tumors exhibit another dimension of complexity: they incorporate a community of recruited, ostensibly normal cells that contribute to the acquisition of hallmark traits by creating the “tumor microenvironment.” Recognition of the widespread applicability of these concepts will increasingly affect the development of new means to treat human cancer.[1]
In an update published in 2011 ("Hallmarks of cancer: the next generation"), Weinberg and Hanahan proposed two new hallmarks: (1) abnormal metabolic pathways and (2) evasion of the immune system, and two enabling characteristics: (1) genome instability, and (2) inflammation.[2]
List of hallmarks
Cancer cells have defects in the control mechanisms that govern how often they divide, and in the feedback systems that regulate these control mechanisms (i.e. defects in homeostasis).
Normal cells grow and divide, but have many controls on that growth. They only grow when stimulated by growth factors. If they are damaged, a molecular brake stops them from dividing until they are repaired. If they can't be repaired, they commit programmed cell death (apoptosis). They can only divide a limited number of times. They are part of a tissue structure, and remain where they belong. They need a blood supply to grow.
All these mechanisms must be overcome in order for a cell to develop into a cancer. Each mechanism is controlled by several proteins. A critical protein must malfunction in each of those mechanisms. These proteins become non-functional or malfunctioning when the DNA sequence of their genes is damaged through acquired or somatic mutations (mutations that are not inherited but occur after conception). This occurs in a series of steps, which Hanahan and Weinberg refer to as hallmarks.
| Capability | Simple analogy |
|---|---|
| Self-sufficiency in growth signals | "accelerator pedal stuck on" |
| Insensitivity to anti-growth signals | "brakes don't work" |
| Evading apoptosis | won't die when the body normally would kill the defective cell |
| Limitless replicative potential | infinite generations of descendants |
| Sustained angiogenesis | telling the body to give it a blood supply |
| Tissue invasion and metastasis | migrating and spreading to other organs and tissues |
Self-sufficiency in growth signals
- Cancer cells do not need stimulation from external signals (in the form of growth factors) to multiply.
Typically, cells of the body require hormones and other molecules that act as signals for them to grow and divide. Cancer cells, however, have the ability to grow without these external signals. There are multiple ways in which cancer cells can do this: by producing these signals themselves, known as autocrine signalling; by permanently activating the signalling pathways that respond to these signals; or by destroying 'off switches' that prevents excessive growth from these signals (negative feedback). In addition, cell division in normal, non-cancerous cells is tightly controlled. In cancer cells, these processes are deregulated because the proteins that control them are altered, leading to increased growth and cell division within the tumor.[4][5]
Insensitivity to anti-growth signals
- Cancer cells are generally resistant to growth-preventing signals from their neighbours.
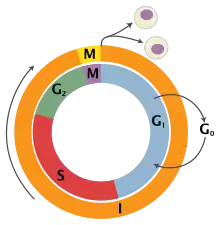

To tightly control cell division, cells have processes within them that prevent cell growth and division. These processes are orchestrated by proteins known as tumor suppressor genes. These genes take information from the cell to ensure that it is ready to divide, and will halt division if not (when the DNA is damaged, for example). In cancer, these tumour suppressor proteins are altered so that they don't effectively prevent cell division, even when the cell has severe abnormalities. Another way cells prevent over-division is that normal cells will also stop dividing when the cells fill up the space they are in and touch other cells; known as contact inhibition. Cancer cells do not have contact inhibition, and so will continue to grow and divide, regardless of their surroundings.[4][6]
Evading programmed cell death
- Apoptosis is a form of programmed cell death (cell suicide), the mechanism by which cells are programmed to die in the event they become damaged. Cancer cells are characteristically able to bypass this mechanism.
Cells have the ability to 'self-destruct'; a process known as apoptosis. This is required for organisms to grow and develop properly, for maintaining tissues of the body, and is also initiated when a cell is damaged or infected. Cancer cells, however, lose this ability; even though cells may become grossly abnormal, they do not undergo apoptosis. The cancer cells may do this by altering the mechanisms that detect the damage or abnormalities. This means that proper signaling cannot occur, thus apoptosis cannot activate. They may also have defects in the downstream signaling itself, or the proteins involved in apoptosis, each of which will also prevent proper apoptosis.[4][7]
Limitless replicative potential
- Non-cancer cells die after a certain number of divisions. Cancer cells escape this limit and are apparently capable of indefinite growth and division (immortality). But those immortal cells have damaged chromosomes, which can become cancerous.
Cells of the body don't normally have the ability to divide indefinitely. They have a limited number of divisions before the cells become unable to divide (senescence), or die (crisis). The cause of these barriers is primarily due to the DNA at the end of chromosomes, known as telomeres. Telomeric DNA shortens with every cell division, until it becomes so short it activates senescence, so the cell stops dividing. Cancer cells bypass this barrier by manipulating enzymes (telomerase) to increase the length of telomeres. Thus, they can divide indefinitely, without initiating senescence.[4][8]
Mammalian cells have an intrinsic program, the Hayflick limit, that limits their multiplication to about 60–70 doublings, at which point they reach a stage of senescence.
This limit can be overcome by disabling their pRB and p53 tumor suppressor proteins, which allows them to continue doubling until they reach a stage called crisis, with apoptosis, karyotypic disarray, and the occasional (10−7) emergence of an immortalized cell that can double without limit. Most tumor cells are immortalized.
The counting device for cell doublings is the telomere, which decreases in size (loses nucleotides at the ends of chromosomes) during each cell cycle. About 85% of cancers upregulate telomerase to extend their telomeres and the remaining 15% use a method called the Alternative Lengthening of Telomeres.[9]
Sustained angiogenesis
- Angiogenesis is the process by which new blood vessels are formed. Cancer cells appear to be able to kickstart this process, ensuring that such cells receive a continual supply of oxygen and other nutrients.
Normal tissues of the body have blood vessels running through them that deliver oxygen from the lungs. Cells must be close to the blood vessels to get enough oxygen for them to survive. New blood vessels are formed during the development of embryos, during wound repair and during the female reproductive cycle. An expanding tumour requires new blood vessels to deliver adequate oxygen to the cancer cells, and thus exploits these normal physiological processes for its benefit. To do this, the cancer cells acquire the ability to orchestrate production of new vasculature by activating the 'angiogenic switch'. In doing so, they control non-cancerous cells that are present in the tumor that can form blood vessels by reducing the production of factors that inhibit blood vessel production, and increasing the production of factors that promote blood vessel formation.[4][10]
Tissue invasion and metastasis
- Cancer cells can break away from their site or organ of origin to invade surrounding tissue and spread (metastasize) to distant body parts.
One of the most well known properties of cancer cells is their ability to invade neighboring tissues. It is what dictates whether the tumor is benign or malignant, and is the property which enables their dissemination around the body. The cancer cells have to undergo a multitude of changes in order for them to acquire the ability to metastasize, in a multistep process that starts with local invasion of the cells into the surrounding tissues. They then have to invade blood vessels, survive in the harsh environment of the circulatory system, exit this system and then start dividing in the new tissue.[4][11]
Updates
In his 2010 NCRI conference talk, Hanahan proposed two new emerging hallmarks and two enabling characteristics. These were later codified in an updated review article entitled "Hallmarks of cancer: the next generation."[2]
Deregulated metabolism
Most cancer cells use alternative metabolic pathways to generate energy, a fact appreciated since the early twentieth century with the postulation of the Warburg hypothesis,[12][13] but only now gaining renewed research interest.[14] Cancer cells exhibiting the Warburg effect upregulate glycolysis and lactic acid fermentation in the cytosol and prevent mitochondria from completing normal aerobic respiration (oxidation of pyruvate, the citric acid cycle, and the electron transport chain). Instead of completely oxidizing glucose to produce as much ATP as possible, cancer cells would rather convert pyruvate into the building blocks for more cells. In fact, the low ATP:ADP ratio caused by this effect likely contributes to the deactivation of mitochondria. Mitochondrial membrane potential is hyperpolarized to prevent voltage-sensitive permeability transition pores (PTP) from triggering of apoptosis.[15][16]
The ketogenic diet is being investigated as an adjuvant therapy for some cancers,[17][18][19] including glioma,[20][21] because of cancer's inefficiency in metabolizing ketone bodies.
Evading the immune system
Despite cancer cells causing increased inflammation and angiogenesis, they also appear to be able to avoid interaction with the body's immune system via a loss of interleukin-33. (See cancer immunology)
Enabling Characteristics
The updated paper also identified two enabling characteristics. These are labeled as such since their acquisition leads to the development of the hypothesized "hallmarks"
Genome instability
Cancer cells generally have severe chromosomal abnormalities which worsen as the disease progresses. HeLa cells, for example, are extremely prolific and have tetraploidy 12, trisomy 6, 8, and 17, and a modal chromosome number of 82 (rather than the normal diploid number of 46).[22] Small genetic mutations are most likely what begin tumorigenesis, but once cells begin the breakage-fusion-bridge (BFB) cycle, they are able to mutate at much faster rates. (See genome instability)
Inflammation
Recent discoveries have highlighted the role of local chronic inflammation in inducing many types of cancer. Inflammation leads to angiogenesis and more of an immune response. The degradation of extracellular matrix necessary to form new blood vessels increases the odds of metastasis. (See inflammation in cancer)
Criticisms
An article in Nature Reviews Cancer in 2010 pointed out that five of the 'hallmarks' were also characteristic of benign tumours.[23] The only hallmark of malignant disease was its ability to invade and metastasize.[23]
An article in the Journal of Biosciences in 2013 argued that original data for most of these hallmarks is lacking.[24] It argued that cancer is a tissue-level disease and these cellular-level hallmarks are misleading.
Notes and references
- Hanahan D, Weinberg RA (January 2000). "The Hallmarks of Cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9. PMID 10647931.
- Hanahan, D.; Weinberg, R. A. (2011). "Hallmarks of Cancer: The Next Generation". Cell. 144 (5): 646–674. doi:10.1016/j.cell.2011.02.013. PMID 21376230.
- Cell 100:59
- Hanahan, D; Weinberg, RA (4 March 2011). "Hallmarks of cancer: the next generation". Cell. 144 (5): 646–74. doi:10.1016/j.cell.2011.02.013. PMID 21376230.
- Evan, GI; Vousden, KH (17 May 2001). "Proliferation, cell cycle and apoptosis in cancer". Nature. 411 (6835): 342–8. Bibcode:2001Natur.411..342E. doi:10.1038/35077213. PMID 11357141. S2CID 4414024.
- McClatchey, AI; Yap, AS (October 2012). "Contact inhibition (of proliferation) redux". Current Opinion in Cell Biology. 24 (5): 685–94. doi:10.1016/j.ceb.2012.06.009. PMID 22835462.
- Elmore, S (June 2007). "Apoptosis: a review of programmed cell death". Toxicologic Pathology. 35 (4): 495–516. doi:10.1080/01926230701320337. PMC 2117903. PMID 17562483.
- Greenberg, RA (March 2005). "Telomeres, crisis and cancer". Current Molecular Medicine. 5 (2): 213–8. doi:10.2174/1566524053586590. PMID 15974875.
- Cesare, Anthony J.; Reddel, Roger R. (2010). "Alternative lengthening of telomeres: Models, mechanisms and implications". Nature Reviews Genetics. 11 (5): 319–330. doi:10.1038/nrg2763. PMID 20351727. S2CID 19224032.
- Bergers, G; Benjamin, LE (June 2003). "Tumorigenesis and the angiogenic switch". Nature Reviews. Cancer. 3 (6): 401–10. doi:10.1038/nrc1093. PMID 12778130. S2CID 11096398.
- van Zijl, F; Krupitza, G; Mikulits, W (2011). "Initial steps of metastasis: cell invasion and endothelial transmigration". Mutation Research. 728 (1–2): 23–34. doi:10.1016/j.mrrev.2011.05.002. PMC 4028085. PMID 21605699.
- Alfarouk, KO; Verduzco, D; Rauch, C; Muddathir, AK; Adil, HH; Elhassan, GO; Ibrahim, ME; David Polo Orozco, J; Cardone, RA; Reshkin, SJ; Harguindey, S (2014). "Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question". Oncoscience. 1 (12): 777–802. doi:10.18632/oncoscience.109. PMC 4303887. PMID 25621294.
- O. Warburg, K. Posener, E. Negelein: "Ueber den Stoffwechsel der Tumoren" Biochemische Zeitschrift, 152, pp. 319–344, 1924. (German). Reprinted in English in the book On metabolism of tumors by O. Warburg, Publisher: Constable, London, 1930.
- "Targeting tumour metabolism". Nature Reviews Drug Discovery. 9 (7): 503–504. 2010. doi:10.1038/nrd3215. ISSN 1474-1776. PMID 20592733. S2CID 7521218.
- Forrest MD. "Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy". bioRxiv 10.1101/025197.
- Gottlieb E, Armour SM, Harris MH, Thompson CB (2003). "Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis". Cell Death Differ. 10 (6): 709–717. doi:10.1038/sj.cdd.4401231. PMID 12761579.
- Schwartz, L; Supuran, CT; Alfarouk, KO (2017). "The Warburg Effect and the Hallmarks of Cancer". Anti-Cancer Agents in Medicinal Chemistry. 17 (2): 164–170. doi:10.2174/1871520616666161031143301. PMID 27804847.
- Barañano KW, Hartman AL (2008). "The ketogenic diet: uses in epilepsy and other neurologic illnesses". Curr Treat Options Neurol. 10 (6): 410–9. doi:10.1007/s11940-008-0043-8. PMC 2898565. PMID 18990309.
- Allen BG, Bhatia SK, Anderson CM, et al. (October 2011). "Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism". Redox Biol. 2C (3): 327–337. doi:10.1016/j.eplepsyres.2011.09.022. PMID 22019313. S2CID 20445641.
- Schwartz, L; Seyfried, T; Alfarouk, KO; Da Veiga Moreira, J; Fais, S (April 2017). "Out of Warburg effect: An effective cancer treatment targeting the tumor specific metabolism and dysregulated pH". Seminars in Cancer Biology. 43: 134–138. doi:10.1016/j.semcancer.2017.01.005. PMID 28122260.
- Scheck AC, Abdelwahab MG, Fenton KE, Stafford P (October 2011). "The ketogenic diet for the treatment of glioma: insights from genetic profiling". Epilepsy Res. 100 (3): 327–37. doi:10.1016/j.eplepsyres.2011.09.022. PMID 22019313. S2CID 20445641.
- "HeLa nuclear extract lysate (ab14655)". abcam.
- Lazebnik Y (April 2010). "What are the hallmarks of cancer?". Nat. Rev. Cancer. 10 (4): 232–3. doi:10.1038/nrc2827. PMID 20355252. S2CID 8862667.
- Sonnenschein C, Soto AM (September 2013). "The aging of the 2000 and 2011 Hallmarks of Cancer reviews: A critique" (PDF). J. Biosci. 38 (3): 651–63. doi:10.1007/s12038-013-9335-6. PMC 3882065. PMID 23938395.