Tumor mutational burden
Tumour mutational burden (abbreviated as TMB) is a genetic characteristic of tumorous tissue that can be informative to cancer research and treatment. It is defined as the number of non-inherited mutations per million bases (Mb) of investigated genomic sequence,[1] and its measurement has been enabled by next generation sequencing. TMB has shown potential as a predictive biomarker with several applications, including associations reported between different TMB levels and patient response to immune checkpoint inhibitor (ICI) therapy in a variety of cancers.[2]
While both TMB and mutational signatures give us critical information about cancer behaviour, they have different definitions. TMB is defined as the number of somatic mutations/megabase whereas mutational signatures are distinct mutational patterns of single base substitutions, double base substitutions, or small insertions and deletions in tumors.[3] For instance, COSMIC single base substitution signature 1 is characterized by the enzymatic deamination of cytosine to thymine and has been associated with age of an individual.[3]
Scientists postulate that high TMB is associated with an increased amount of neoantigens, which are tumour specific markers displayed by cells.[4] An increase in these antigens may then lead to increased detection of cancer cells by the immune system and more robust activation of cytotoxic T-lymphocytes. Activation of T-cells is further regulated by immune checkpoints that can be displayed by cancer cells, thus treatment with ICIs can lead to improved patient survival.[5]
On June 16, 2020 the U.S. Food and Drug Administration expanded the approval of the immunotherapy drug pembrolizumab to treat any advanced solid-tumor cancers with a TMB greater than 10 mutations per Mb and continued growth following prior treatments.[6] This marks the first time that the FDA has approved a drug with its use based on TMB measurements.[7]

Importance
TMB as a Biomarker
One survival mechanism in tumors is to increase the expression of immune checkpoint molecules that can bind to tumor-specific T-cells and inactivate them, so that the tumor cells cannot be detected and killed.[8] ICIs have been shown to improve patients’ response and the survival rates as they help the immune system to target tumor cells.[1][8] However, there is a variation in response to ICIs among patients and it is crucial to know which patients can benefit from ICI therapy.[1] The expression of PD-L1 (programmed death-ligand 1; one of the immune checkpoints) has been demonstrated to be a good biomarker of PD-L1 blockade therapy in some cancers.[8] However, there is a need for better biomarkers as there are some predictive errors with PD-L1 expression.[8] Studies on TMB have illustrated that there is an association between patients’ outcome (of ICI therapy) and the TMB value.[1] It has been proposed that TMB can be used as a predictive marker of response in ICI therapy across many cancer types.[8] Also, TMB can be helpful to identify individuals that can benefit from ICI therapy with cancers that generally have low TMB values.[8] Furthermore, it has been shown that tumors with higher TMB values usually result in a higher number of neoantigens, the antigens that are presented on the tumor cells surface that are usually a result of missense mutations.[8] So, TMB can be a good estimator of neoantigen load and can help find the patients who can benefit from ICI therapy by increasing the chance of detecting the neoantigens.[8] However, it is important to note that different sequencing platforms and bioinformatics pipelines have been used to estimate TMB and it is important to harmonize TMB quantification protocols and procedures before it can be used as a reliable biomarker.[1][9] There have been some efforts to standardize these methods.[1]
Treatment Response
TMB has been found to correlate with patient response to therapies such as immune checkpoint inhibitors (ICIs). An analysis of a large cohort of patients receiving ICI therapy revealed that higher TMB levels (≥ 20 mutations/Mb) corresponded to a 58% response rate to ICIs while lower TMB levels (<20 mutations/Mb) reduced response to 20%.[10] Researchers could also show a significant correlation between treatment response rate and TMB level in patients treated with anti-PD-1 or anit-PD-L1 (types of ICIs).[11] Additionally, it has been reported that when ICIs were the only treatments used by patients, 55% of the differences in the objective response rate across cancer types were explained by TMB.[11]
Patient Prognosis
Associations have been reported between TMB and patient outcome in a variety of cancers. In one study, scientists observed differences in survival rates, with high TMB individuals having a median progression-free survival of 12.8 months and a median overall survival not reached by the time of publication, compared to 3.3 months and 16.3 months respectively for individuals with lower TMB.[10] Another study examining patients who had not received ICI therapy found that intermediate levels of TMB (>5 and <20 mutations/Mb) correlate with significantly decreased survival, likely as a result of the accumulation of mutations in oncogenes.[5] This relationship does not appear to be significantly disparate across different tissues types and is only modestly affected by corrections for confounders such as smoking, sex, age, and ethnicity.[5] This suggests that TMB is both an independent and reliable indicator of poor patient outcomes in the absence of ICI therapy.[5] Interestingly, very high levels of TMB (≥ 50 mutations/Mb) were reported to correlate with increased survival, giving an overall parabolic shape to the trend.[5] While this association is still under investigation, it has been hypothesized that the decreased risk of death under very high TMB could result from reduced cell viability due to genetic instability or increased production of neoantigens recognized by the immune system.[5]
TMB in different cancers
There is a large variation in TMB values across different cancer types as the number of somatic mutations can span from 0.01 to 400 mutations per megabase of genome.[1][8][9] It has been shown that melanoma, NSCLC and other squamous carcinomas have the highest levels of TMB in this order, while leukemias and pediatric tumors have the lowest levels of TMB and other cancers like breast, kidney, and ovary have intermediate TMB values.[8] There is also variation in TMB across different subtypes of different cancers.[8] Due to high variability in TMB across different cancer types and subtypes, it is important to define different cut-offs to have an improved survival prediction and a better treatment decision.[1][8][9] For example, Fernandez et al. showed that TMB can range from 0.03 to 14.13 mutations per megabase (mean=1.23) in TCGA prostate cancer cohort while this range is from 0.04-99.68 mutations per megabase (mean=6.92) in TCGA bladder cancer cohort.[12] A recent study illustrated that different cut-offs are needed for different cancer types to find the patients who can benefit from ICI therapy.[1] In addition, it is crucial to understand that usually there are different clusters of cells in a tumor, known as tumor heterogeneity, that can affect TMB and consequently the response to ICIs.[8] Another factor that can affect TMB is whether the source of the sample is primary or metastatic tissue.[13] Most metastatic samples have been shown to be monoclonal (i.e. there is only one cluster of cells in the tumor), while primary tumors usually consist of a higher number of clusters and have higher overall genetic diversity (more heterogeneous).[13] Scientists have shown that metastatic tumors usually have a higher TMB level compared to primary tumors and this can be due to monoclonal nature of metastatic lesions.[13]
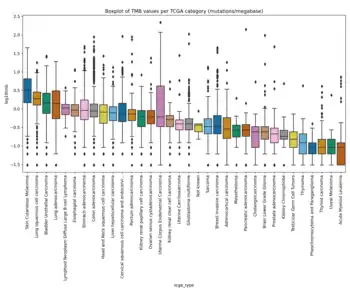
TMB calculation
There exists disparities between how TMB is calculated in clinical and research settings.[14] Broadly, whole genome sequencing, whole exome sequencing, and panel based approaches can be used to help to calculate TMB.[14] Studies of TMB from research perspectives typically incorporate whole exome sequencing, and occasionally whole genome sequencing within their workflows while clinical applications use panel sequencing to estimate TMB primarily for their comparatively quicker speed and low cost.[14] Within panel based approaches, different strategies to calculate TMB have been adopted.[14] For instance, consider MSK-IMPACT developed by the Memorial Sloan Kettering Cancer Center and F1CDx developed by Foundation Medicine.[15][16] F1CDx utilizes tumor-only sequencing strategy while MSK-IMPACT requires sequencing of both the tumor and its matched normal sample. Additionally, F1CDx counts synonymous mutations while excluding hotspot driver mutations.[15] MSK-IMPACT calculates TMB with similar filtering criteria to those used in whole exome sequencing, considering both synonymous mutations and hotspot driver mutations.[16] Ensembles of targeted panels and whole exome sequencing panels have been recommended for optimal results.[17] As an approach that is potentially more expedient and cost effective than sequencing, TMB can be calculated directly from H&E stained pathology images using deep learning.[18]
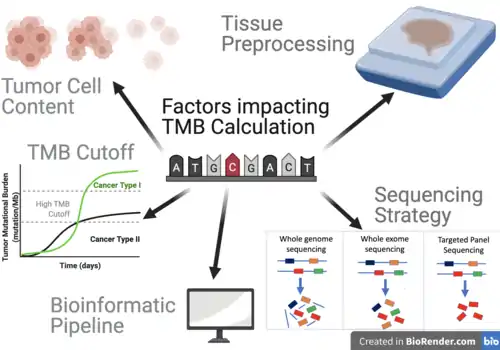
Factors that Influence TMB Calculation
Overall, 5 primary factors have been identified to influence TMB calculations.[19]
Tumor Cell Content and Sequencing Coverage
Greater tumor cell content and sequencing coverage play a key role in the quality of TMB data.[19] For instance, targeted panels may enable deeper sequencing compared to whole exome sequencing, enabling higher sensitivity, that have been shown to perform well even when tumor cell content is low (defined as <10%).[19] Targeted panels have shown to enable much greater coverage than in whole exome sequencing.[19] For example, one recent study reached a mean sequencing coverage across all tumor samples of 744× when using the MSK-IMPACT panel, while the WES led to a mean target coverage of 232× in tumor sequences.[20]
Tissue Preprocessing
Typically, tumor tissues are fixated in formalin to preserve tissue and cellular morphology in the formalin-fixed paraffin-embedded (FFPE) protocols.[21] While FFPE offers a cost-effective method to store tissues for long durations of time, limitations must be considered as to how it will affect TMB calculations.[21] One limitation of this method is that it induces the formation of various crosslinks, whereby strands of DNA become covalently bound to each other, which may consequently lead to deamination of cytosine bases.[19] Cytosine deamination is the major cause of baseline noise in Next Generation Sequencing, leading to the most prevalent sequence artifacts in FFPE (C:G > T:A).[19] This may generate artefacts that must be removed in the downstream pipeline.
Sequencing Strategy
Different sequencing strategies enable different number of genes to be included in the calculation of TMB (with WGS and WES approaches allowing a greater quantity of genes to be analyzed). While panel based approaches analyze comparatively fewer genes than other strategies, one advantage of panel based sequencing is that genes of interest can be covered in much greater sequencing depths, and rare variants can possibly be identified.[19] The panel sizes vary across panels with 468 genes in the MSK-IMPACT panel, 315 genes in the Foundation Medicine panel, and 409 genes in the Life Technologies panel.[19] As panel sizes are smaller, uncertainty associated with TMB estimation becomes greater, with coefficient of variance increases rapidly when the size of the targeted panels is less than 1 Mb.[21]
Bioinformatics Pipeline
In most calculations of TMB, synonymous variants and germline variants are filtered out as they are unlikely to be directly involved in creating neoantigens.[19] However, some pipelines maintain synonymous variants.[21] To account for germline variants, ideally sequencing would have been performed on a matched non-tumor sample from each patient.[21] However, in a clinical practice, the availability of this matched sample may vary across different institutions and diverse organizational factors, and data unavailability may inhibit germline variants to be filtered.[21] The choice of variant callers and other software in the downstream analyses may also affect how TMB is ultimately calculated.[21] TMB can be calculated directly from histopathology images using a multiscale deep learning pipeline, avoiding the need for sequencing and variant calling.[18]
Cut-offs
Different studies have assigned different cut-offs to delineate between high and low TMB status.[19] In the lung, the median TMB across more than 18,000 lung cancer cases was 7.2 mutations/Mb, with approximately 12% of the patients showing more than 20 mutations/Mb.[21] The authors identified a tumor mutational burden greater than or equal to 10 mutations/Mb as the optimal cut-off to benefit from combination immunotherapy.[21] However, in other cancer types, high TMB status has been classified as >20 mutations/Mb.[5]
Issues and future directions
One approved biomarker of ICI therapy is PD-L1 expression, but the predictive power of this biomarker is affected by factors such as assay interpretation and lack of standard methods.[8] TMB is also affected by these factors in addition to accessibility issues.[8] Biological factors like specimen type and cancer type as well as technical factors like sequencing technology can affect evaluation of TMB.[1] Thus, it is necessary to harmonize evaluation methods and there are still so many factors that can complicate this task.[1][8] For example, gene fusions and post-translational changes in proteins contribute to tumor behaviour and consequently response to therapy while these factors are not considered in TMB estimation.[8] In addition, currently all mutations have the same weight in TMB calculation, while they can have very different effects on proteins and pathways activity.[8] Furthermore, there is still no good answer to the question of how mutations in genes that are known to influence ICI therapy should be treated in TMB evaluation.[8] It is also important to note that TMB is highly variable across cancer types and subtypes and different studies are being conducted to find distinct TMB thresholds.[8]
Some studies argue that to have better prediction of response to ICI therapy, TMB should be used as a complementary marker with other biomarkers such as PD-L1.[8] Other studies have shown that a combination of TMB and neoantigen load can be used as a biomarker to predict survival in patients with melanoma who received adaptive T cell transfer therapy.[8] Since TMB is a relatively new biomarker, there is still a need to perform more studies and many labs are being focused on different aspects of this biomarker.[8][9]
References
- Merino DM, McShane LM, Fabrizio D, Funari V, Chen S, White JR, et al. (2020). "Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project". J Immunother Cancer. 8 (1): e000147. doi:10.1136/jitc-2019-000147. PMC 7174078. PMID 32217756.
- Kim JY, Kronbichler A, Eisenhut M, Hong SH, van der Vliet HJ, Kang J, et al. (2019). "Tumor Mutational Burden and Efficacy of Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis". Cancers. 11 (11): 1798. doi:10.3390/cancers11111798. PMC 6895916. PMID 31731749.
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AW, Wu Y, et al. (2020). "The repertoire of mutational signatures in human cancers". Nature. 578 (7793): 94–101. Bibcode:2020Natur.578...94A. doi:10.1038/s41586-020-1943-3. PMC 7054213. PMID 32025018.
- Owada-Ozaki Y, Muto S, Takagi H, Inoue T, Watanabe Y, Fukuhara M, et al. (2018). "Prognostic Impact of Tumour Mutation Burden in Patients with Completely Resected Non-Small Cell Lung Cancer: Brief Report". Journal of Thoracic Oncology. 13 (8): 1217–1221. doi:10.1016/j.jtho.2018.04.003. PMID 29654927. S2CID 4863075.
- Riviere P, Goodman AM, Okamura R, Barkauskas DA, Whitchurch TJ, Lee S, et al. (2020). "High Tumor Mutational Burden Correlates with Longer Survival in Immunotherapy-Naïve Patients with Diverse Cancers". Molecular Cancer Therapeutics. 19 (10): 2139–2145. doi:10.1158/1535-7163.MCT-20-0161. PMC 7541603. PMID 32747422.
- "FDA approves pembrolizumab for adults and children with TMB-H solid tumors". U.S. Food and Drug Administration. 2020. Retrieved February 18, 2021.
- "FDA Approves First Drug for Cancers with a High Tumor Mutational Burden". American Cancer Society. 2020. Retrieved February 18, 2021.
- Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. (2019). "Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic". Ann Oncol. 30 (1): 44–56. doi:10.1093/annonc/mdy495. PMC 6336005. PMID 30395155.
- Addeo A, Banna GL, Weiss GJ (2019). "Tumor Mutation Burden-From Hopes to Doubts". JAMA Oncol. 5 (7): 934–935. doi:10.1001/jamaoncol.2019.0626. PMID 31145420. S2CID 169038765.
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. (2017). "Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers". Molecular Cancer Therapeutics. 16 (11): 2598–2608. doi:10.1158/1535-7163.MCT-17-0386. PMC 5670009. PMID 28835386.
- Yarchoan M, Hopkins A, Jaffee EM (2017). "Tumour Mutational Burden and Response Rate to PD-1 Inhibition". N Engl J Med. 377 (25): 2500–2501. doi:10.1056/NEJMc1713444. PMC 6549688. PMID 29262275.
- Fernandez EM, Eng K, Beg S, Beltran H, Faltas BM, Mosquera JM, et al. (2019). "Cancer-Specific Thresholds Adjust for Whole Exome Sequencing-based Tumor Mutational Burden Distribution". JCO Precis Oncol. 3 (3): 1–12. doi:10.1200/PO.18.00400. PMC 6716608. PMID 31475242.
- Schnidrig D, Turajlic S, Litchfield K (2019). "Tumor mutational burden: primary versus metastatic tissue creates systematic bias". IOTECH. 4: 8–14. doi:10.1016/j.iotech.2019.11.003. PMC 9216665. PMID 35755001.
- Xu Z, Dai J, Wang D, Lu H, Dai H, Ye H, et al. (2019). "Assessment of tumor mutation burden calculation from gene panel sequencing data". OncoTargets Ther. 12: 3401–9. doi:10.2147/OTT.S196638. PMC 6510391. PMID 31123404.
- Büttner R, Longshore JW, López-Ríos F, Merkelbach-Bruse S, Normanno N, Rouleau E, et al. (2019). "Implementing TMB measurement in clinical practice: considerations on assay requirements". ESMO Open. 4 (1): e000442. doi:10.1136/esmoopen-2018-000442. PMC 6350758. PMID 30792906.
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. (2015). "Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology". J Mol Diagn JMD. 17 (3): 251–64. doi:10.1016/j.jmoldx.2014.12.006. PMC 5808190. PMID 25801821.
- Shao C, Li G, Huang L, Pruitt S, Castellanos E, Frampton G, et al. (2020). "Prevalence of High Tumor Mutational Burden and Association With Survival in Patients With Less Common Solid Tumors". JAMA Netw Open. 3 (10): e2025109. doi:10.1001/jamanetworkopen.2020.25109. PMC 7596577. PMID 33119110.
- Jain, Mika S.; Massoud, Tarik F. (2020). "Predicting tumour mutational burden from histopathological images using multiscale deep learning". Nature Machine Intelligence. 2 (6): 356–362. doi:10.1038/s42256-020-0190-5. ISSN 2522-5839. S2CID 220510782.
- Meléndez B, Van Campenhout C, Rorive S, Remmelink M, Salmon I, D'Haene N (2018). "Methods of measurement for tumor mutational burden in tumor tissue". Transl Lung Cancer Res. 7 (6): 661–7. doi:10.21037/tlcr.2018.08.02. PMC 6249625. PMID 30505710.
- Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. (2018). "Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing". J Clin Oncol. 36 (7): 633–41. doi:10.1200/JCO.2017.75.3384. PMC 6075848. PMID 29337640.
- Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. (2017). "Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden". Genome Med. 9 (1): 34. doi:10.1186/s13073-017-0424-2. PMC 5395719. PMID 28420421.