Vestibulocerebellar syndrome
Vestibulocerebellar syndrome, also known as vestibulocerebellar ataxia, is a progressive neurological disorder that causes a variety of medical problems. Initially symptoms present as periodic attacks of abnormal eye movements but may intensify to longer-lasting motor incapacity. The disorder has been localized to the vestibulocerebellum, specifically the flocculonodular lobe.[1] Symptoms of vestibulocerebellar syndrome may appear in early childhood but the full onset of neurological symptoms including nystagmus (involuntary eye movement), ataxia (loss of voluntary muscle coordination), and tinnitus (perception of sound in the absence of external stimulation) does not occur until early adulthood.[2] To date, vestibulocerebellar syndrome has only been identified in three families but has affected multiple generations within them. Based on the familial pedigrees it has been characterized as an autosomal dominant disorder, although the exact genetic locus has not been identified.[2][3] It has been found to be genetically distinct from other seemingly similar forms of neurological syndromes such as episodic ataxia types 1 and 2. Due to its rarity, however, little is known about specific details of the pathology or long-term treatment options.[2] There is currently no cure for vestibulocerebellar syndrome, although some drug therapies have been effective in alleviating particular symptoms of the disorder.
| Vestibulocerebellar syndrome | |
|---|---|
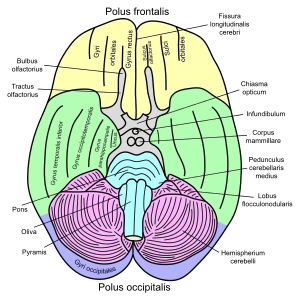 | |
| Basal view of the human brain including the cerebellum |
Symptoms
The symptoms of vestibulocerebellar syndrome vary among patients but are typically a unique combination of ocular abnormalities including nystagmus, poor or absent smooth pursuit (ability of the eyes to follow a moving object), strabismus (misalignment of the eyes), diplopia (double vision), oscillopsia (the sensation that stationary objects in the visual field are oscillating) and abnormal vestibulo-ocular reflex (reflex eye adjustment to stabilize gaze during head movement).[4] Gaze-paretic nystagmus, one of the most common symptoms among patients results in poor gaze-holding due to neuron integrator dysfunction. Rebound nystagmus is also frequently found in conjunction with gaze-paretic nystagmus and is characteristic of cerebellar malfunction.[5] These abnormal eye movements are often the earliest indicators of the disorder and may appear during childhood. The time of full onset of symptoms, including motor abnormalities, ranges from age 30 to age 60. Initially, symptoms present as isolated episodic attacks but occur at increasing frequency over time and may eventually become a permanent condition.[2] In conjunction with eye abnormalities patients also present with periodic attacks of vertigo, tinnitus, and ataxia that are associated with sudden changes in head position.[3] Attacks were seen to vary in duration from a few minutes to months in different individuals and were often accompanied by nausea and vomiting.[6]
During a typical attack, patients reported having ataxic gait with a tendency to fall to either side while lacking the ability to walk heel-to-toe.[6] With more severe attacks, patients had to sit down due to extreme unsteadiness. Fine motor abilities, such as writing and buttoning clothes, became impaired during an attack. However, speech remained unaffected. Attacks did not cause a loss of consciousness nor did they disturb mental activity.[6] Once the attack ended, oscillopsia faded and the intensity of nystagmus decreased.[5] Although these attacks are similar to episodic ataxia, patients with vestibulocerebellar syndrome do not completely lose motor control in arms and legs or experience dysarthria (poor speech articulation), as patients with episodic ataxia do.[7] The disturbances to vestibular function during periodic attacks are the primary distinction between vestibulocerebellar syndrome and other similar neurological syndromes. These conditions do not consistently cause the symptoms of dizziness and ocular impairment that have been localized to the vestibulocerebellum, leading researchers to characterize vestibulocerebellar syndrome as a distinct disorder.
Causes

Vestibulocerebellar syndrome is caused by a failure in the function of the flocculus of the vestibulocerebellum, one of the three main divisions of the cerebellum. Generally, the cerebellum is responsible for regulating motor commands. The main function of the vestibulocerebellum is to receive sensory input from the vestibular nuclei in the brainstem and to regulate equilibrium, balance, and the vestibulo-ocular reflex accordingly. The vestibulo-ocular reflex, one of the primary areas affected by vestibulocerebellar syndrome, is responsible for counterrotating the eyes in response to head movements. This allows gaze to stay fixed on a specific point. A failure in this reflex results in a variety of eye movement abnormalities, such as those exhibited in vestibulocerebellar syndrome.[8]
Vestibulocerebellar syndrome has been categorized as an autosomal dominant neurological disorder although the specific effect on the vestibulocerebellum is unknown. It is possible that inheritance causes abnormalities in either the flocculus or in structures that project into the flocculus to maintain stability of the retinal image of stationary or moving visual objects.[4] Pathological symptoms of the disorder may appear within the first 1–2 years of life although time of onset varies greatly among patients. The severity of symptoms typically progresses with age. The exact cause of the disorder and its pathogenic effect on the flocculus is unknown. A single genetic locus, however, critical in early eye movement control pathways on chromosome 13q31-q33 has been discovered. This locus may be involved in some of the ocular abnormalities that occur in affected individuals. Chromosome 13q31-q33, however, has not been seen to correspond to any known existing gene or locus responsible for congenital nystagmus, one of the primary symptoms of vestibulocerebellar syndrome, or for better-understood cerebellar ataxias.[4]
Vestibulocerebellar syndrome shares clinical similarities with autosomal dominant ataxias, particularly episodic ataxia types 1 and 2. These similarities include gaze-evoked and rebound nystagmus and vertigo. Furthermore, the symptoms appear to progress over time.[9] The molecular basis of many of these other disorders has been thoroughly established and in some cases a genetic locus has been identified. Despite the similarities between symptoms of episodic ataxia and vestibulocerebellar syndrome, studies of affected individuals have shown that the disorder is genetically distinct from these other similar neurological conditions. To date, the molecular basis of vestibulocerebellar syndrome remains undefined.[2]
Management
Attacks of vestibulocerebellar syndrome may be triggered by a sudden change in head position, fatigue, or by being in an environment of fast-moving objects. These attacks may be alleviated by lying quietly with eyes closed for fifteen to thirty minutes. Lying down stabilizes the head in a fixed position while closing the eyes removes the unstable sensory input responsible for the dizziness. Treatment is presently focused on addressing specific symptoms to alleviate the nausea that often accompanies attacks and to make daily life more manageable. There have been very few studies on the effectiveness of drugs that are used for the management of other ataxias on vestibulocerebellar syndrome.[2] Trials with acetazolamide achieved some success, while amitriptyline hydrochloride was unsuccessful.[2][3] Acetazolamide therapy proved effective in treating episodic vertigo. In trials of patients who have vestibulocerebellar syndrome, acetazolamide either eliminated or significantly decreased the frequency and severity of vertigo episodes.[10] Dramamine (Dimenhydrinate) and antihistamine drugs have also been helpful in decreasing the frequency and severity of attacks.[6]
Prognosis
There is currently no cure for vestibulocerebellar ataxia.[10] When a diagnosis is made in early adulthood on the basis of periodic attacks of vertigo, diplopia, and tinnitus, one can expect recurrent episodes of progressive ataxia later in life.[5] In order to confirm the presence of vestibulocerebellar ataxia, a family examination demonstrating similar symptoms is critical in order to prevent a misdiagnosis.[7] A person with this disorder will find daily tasks increasingly difficult as progressive ataxia develops. Attacks can eventually increase with more frequency and become a permanent condition.
Origin and history
Periodic vestibulocerebellar syndrome has been discovered in several generations of three families with genetic ties to Johnston County, North Carolina.[3] The first was discovered in 1963 by T.W. Farmer and a group of researchers who studied and published a paper on the syndrome. The second case was studied and published in 1984 by Vance et al.[6][11] Individuals in these families were categorized as affected if they exhibited one form of primary criteria (ataxia, marked loss of smooth pursuit, gaze-evoked nystagmus, and impaired vestibulo-ocular reflex suppression) and at least one secondary criteria (mild loss of smooth pursuit, mild gaze-evoked nystagmus, and esophoria or esotropia).[2]
Historically vestibulocerebellar syndrome has been difficult to classify because of the variation in symptoms, severity, and time of onset. During the earlier stages of attacks, members of the third family classified as having vestibulocerebellar syndrome were unaware that other family members experienced the same debilitating symptoms. It was not until after researchers, Vance et al. examined the family history that a diagnosis was made characterizing three generations of the family as affected by vestibulocerebellar syndrome.[11] In addition to these variations, vestibulocerebellar syndrome is also difficult to distinguish from other neurological disorders that result in similar degenerative symptoms such as ataxia and multiple sclerosis.[7]
Research directions
There has been no gene or locus determined to cause vestibulocerebellar syndrome. Genes involved in central nervous system development or maintenance, however, may be considered as candidate genes. As of 2003, research is being done to investigate the potential role of these genes in vestibulocerebellar syndrome. Some possible candidate genes include, SOX21, ZIC2, and TYRP2. Genes that are part of the SOX family are expressed in the developing embryonic brain. Although no individuals have presented with brain abnormalities, it is possible that one of these candidate genes could have a more minor mutation, leading to the symptoms of vestibulocerebellar syndrome. TYRP2, for example, is important in the development of correct pigmentation; general and ocular albinism is associated with nystagmus. Since there is no anatomical correlations with this syndrome, failure of flocculus function is thought to be the cause of the eye movement abnormalities. Researchers surmise, therefore, that there exists a gene critical to establishing early eye movement control pathways.[4]
References
- Theunissen EJ, Huygen PL, Verhagen WI (February 1989). "Familial vestibulocerebellar dysfunction: a new syndrome?". Journal of the Neurological Sciences. 89 (2–3): 149–55. doi:10.1016/0022-510x(89)90016-6. PMID 2926446. S2CID 31498395.
- Damji KF, Allingham RR, Pollock SC, et al. (April 1996). "Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias". Archives of Neurology. 53 (4): 338–44. doi:10.1001/archneur.1996.00550040074016. PMID 8929156.
- Small KW, Pollock SC, Vance JM, Stajich JM, Pericak-Vance M (June 1996). "Ocular motility in North Carolina autosomal dominant ataxia". J Neuroophthalmol. 16 (2): 91–5. doi:10.1097/00041327-199606000-00002. PMID 8797163. S2CID 39897382.
- Ragge NK, Hartley C, Dearlove AM, Walker J, Russell-Eggitt I, Harris CM (January 2003). "Familial vestibulocerebellar disorder maps to chromosome 13q31-q33: a new nystagmus locus". J. Med. Genet. 40 (1): 37–41. doi:10.1136/jmg.40.1.37. PMC 1735258. PMID 12525540.
- Harris CM, Walker J, Shawkat F, Wilson J, Russell-Eggitt I (June 1993). "Eye movements in a familial vestibulocerebellar disorder". Neuropediatrics. 24 (3): 117–22. doi:10.1055/s-2008-1071526. PMID 8355816.
- FARMER TW, MUSTIAN VM (May 1963). "Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations". Arch. Neurol. 8: 471–80. doi:10.1001/archneur.1963.00460050021002. PMID 13944410.
- Farris BK, Smith JL, Ayyar DR (October 1986). "Neuro-ophthalmologic findings in vestibulocerebellar ataxia". Archives of Neurology. 43 (10): 1050–3. doi:10.1001/archneur.1986.00520100056015. PMID 3489454.
- Purves, Dale. Neuroscience. 4th ed. Sunderland, MA: Sinauer, 2008.
- Jen J, Kim GW, Baloh RW (January 2004). "Clinical spectrum of episodic ataxia type 2". Neurology. 62 (1): 17–22. doi:10.1212/01.wnl.0000101675.61074.50. PMID 14718690. S2CID 28147381.
- Baloh RW, Winder A (March 1991). "Acetazolamide-responsive vestibulocerebellar syndrome: clinical and oculographic features". Neurology. 41 (3): 429–33. doi:10.1212/wnl.41.3.429. PMID 2006014. S2CID 45489324.
- Vance, J. M., M. A. Pericak-Vance, C. S. Payne, J. T. Coin, and C. W. Olanow. "Linkage and Genetic Analysis in Adult Onset Periodic Vestibulo-Cerebellar Ataxia: Report of a New Family." American Journal of Human Genetics 36 (1984): 78S