Copolymer
In polymer chemistry, a copolymer is a polymer derived from more than one species of monomer. The polymerization of monomers into copolymers is called copolymerization. Copolymers obtained by copolymerization of two monomer species are sometimes called bipolymers. Those obtained from three and four monomers are called terpolymers and quaterpolymers, respectively.[1]
Commercial copolymers include acrylonitrile butadiene styrene (ABS), styrene/butadiene co-polymer (SBR), nitrile rubber, styrene-acrylonitrile, styrene-isoprene-styrene (SIS) and ethylene-vinyl acetate, all formed by chain-growth polymerization. Another production mechanism is step-growth polymerization, used to produce the nylon-12/6/66 copolymer[2] of nylon 12, nylon 6 and nylon 66, as well as the copolyester family.
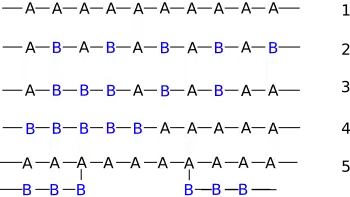
Since a copolymer consists of at least two types of constituent units (also structural units), copolymers can be classified based on how these units are arranged along the chain.[3] Linear copolymers consist of a single main chain, and include alternating copolymers, statistical copolymers and block copolymers. Branched copolymers consist of a single main chain with one or more polymeric side chains, and can be grafted, star shaped or have other architectures.
Reactivity ratios
The reactivity ratio of a growing copolymer chain terminating in a given monomer is the ratio of the reaction rate constant for addition of the same monomer and the rate constant for addition of the other monomer. That is, and , where for example is the rate constant for propagation of a polymer chain ending in monomer 1 (or A) by addition of monomer 2 (or B).[4]



The composition and structural type of the copolymer depend on these reactivity ratios r1 and r2 according to the Mayo–Lewis equation, also called the copolymerization equation or copolymer equation,[5][4] for the relative instantaneous rates of incorporation of the two monomers.

Linear copolymers
Block copolymers


Block copolymers comprise two or more homopolymer subunits linked by covalent bonds. The union of the homopolymer subunits may require an intermediate non-repeating subunit, known as a junction block. Diblock copolymers have two distinct blocks; triblock copolymers have three. Technically, a block is a portion of a macromolecule, comprising many units, that has at least one feature which is not present in the adjacent portions.[1] A possible sequence of repeat units A and B in a triblock copolymer might be ~A-A-A-A-A-A-A-B-B-B-B-B-B-B-A-A-A-A-A~.[6]
Block copolymers are made up of blocks of different polymerized monomers. For example, polystyrene-b-poly(methyl methacrylate) or PS-b-PMMA (where b = block) is usually made by first polymerizing styrene, and then subsequently polymerizing methyl methacrylate (MMA) from the reactive end of the polystyrene chains. This polymer is a "diblock copolymer" because it contains two different chemical blocks. Triblocks, tetrablocks, multiblocks, etc. can also be made. Diblock copolymers are made using living polymerization techniques, such as atom transfer free radical polymerization (ATRP), reversible addition fragmentation chain transfer (RAFT), ring-opening metathesis polymerization (ROMP), and living cationic or living anionic polymerizations.[7] An emerging technique is chain shuttling polymerization.
The synthesis of block copolymers requires that both reactivity ratios are much larger than unity (r1 >> 1, r2 >> 1) under the reaction conditions, so that the terminal monomer unit of a growing chain tends to add a similar unit most of the time.[8]
The "blockiness" of a copolymer is a measure of the adjacency of comonomers vs their statistical distribution. Many or even most synthetic polymers are in fact copolymers, containing about 1-20% of a minority monomer. In such cases, blockiness is undesirable.[9] A block index has been proposed as a quantitative measure of blockiness or deviation from random monomer composition.[10]
Alternating copolymers
An alternating copolymer has regular alternating A and B units, and is often described by the formula: -A-B-A-B-A-B-A-B-A-B-, or -(-A-B-)n-. The molar ratio of each monomer in the polymer is normally close to one, which happens when the reactivity ratios r1 and r2 are close to zero, as can be seen from the Mayo–Lewis equation. For example, in the free-radical copolymerization of styrene maleic anhydride copolymer, r1 = 0.097 and r2 = 0.001,[8] so that most chains ending in styrene add a maleic anhydride unit, and almost all chains ending in maleic anhydride add a styrene unit. This leads to a predominantly alternating structure.
A step-growth copolymer -(-A-A-B-B-)n- formed by the condensation of two bifunctional monomers A–A and B–B is in principle a perfectly alternating copolymer of these two monomers, but is usually considered as a homopolymer of the dimeric repeat unit A-A-B-B.[4] An example is nylon 66 with repeat unit -OC-( CH2)4-CO-NH-(CH2)6-NH-, formed from a dicarboxylic acid monomer and a diamine monomer.
Periodic copolymers
Periodic copolymers have units arranged in a repeating sequence. For two monomers A and B, for example, they might form the repeated pattern (A-B-A-B-B-A-A-A-A-B-B-B)n.
Statistical copolymers
In statistical copolymers the sequence of monomer residues follows a statistical rule. If the probability of finding a given type monomer residue at a particular point in the chain is equal to the mole fraction of that monomer residue in the chain, then the polymer may be referred to as a truly random copolymer[11] (structure 3).
Statistical copolymers are dictated by the reaction kinetics of the two chemically distinct monomer reactants, and are commonly referred to interchangeably as “random” in the polymer literature.[12] As with other types of copolymers, random copolymers can have interesting and commercially desirable properties that blend those of the individual homopolymers. Examples of commercially relevant random copolymers include rubbers made from styrene-butadiene copolymers and resins from styrene-acrylic or methacrylic acid derivatives.[13] Copolymerization is particularly useful in tuning the glass transition temperature, which is important in the operating conditions of polymers; it is assumed that each monomer occupies the same amount of free volume whether it is in a copolymer or homopolymer, so the glass transition temperature (Tg) falls between the values for each homopolymer and is dictated by the mole or mass fraction of each component.[12]
A number of parameters are relevant in the composition of the polymer product; namely, one must consider the reactivity ratio of each component. Reactivity ratios describe whether the monomer reacts preferentially with a segment of the same type or of the other type. For example, a reactivity ratio that is less than one for component 1 indicates that this component reacts with the other type of monomer more readily. Given this information, which is available for a multitude of monomer combinations in the “Wiley Database of Polymer Properties”,[14] the Mayo-Lewis equation can be used to predict the composition of the polymer product for all initial mole fractions of monomer. This equation is derived using the Markov model, which only considers the last segment added as affecting the kinetics of the next addition; the Penultimate Model considers the second-to-last segment as well, but is more complicated than is required for most systems.[15] When both reactivity ratios are less than one, there is an azeotropic point in the Mayo-Lewis plot. At this point, the mole fraction of monomer equals the composition of the component in the polymer.[12]
There are several ways to synthesize random copolymers. The most common synthesis method is free radical polymerization; this is especially useful when the desired properties rely on the composition of the copolymer rather than the molecular weight, since free radical polymerization produces relatively disperse polymer chains. Free radical polymerization is less expensive than other methods, and produces high-molecular weight polymer quickly.[16] Several methods offer better control over dispersity. Anionic polymerization can be used to create random copolymers, but with several caveats: if carbanions of the two components do not have the same stability, only one of the species will add to the other. Additionally, anionic polymerization is expensive and requires very clean reaction conditions, and is therefore difficult to implement on a large scale.[12] Less disperse random copolymers are also synthesized by ″living″ controlled radical polymerization methods, such as atom-transfer radical-polymerization (ATRP), nitroxide mediated radical polymerization (NMP), or Reversible addition−fragmentation chain-transfer polymerization (RAFT). These methods are favored over anionic polymerization because they can be performed in conditions similar to free radical polymerization. The reactions require longer experimentation periods than free radical polymerization, but still achieve reasonable reaction rates.[17]
Stereoblock copolymers
In stereoblock copolymers the blocks or units differ only in the tacticity of the monomers.
Gradient copolymers
In gradient copolymers the monomer composition changes gradually along the chain.
Branched copolymers
There are a variety of architectures possible for nonlinear copolymers. Beyond grafted and star polymers discussed below, other common types of branched copolymers include brush copolymers and comb copolymers.
Graft copolymers
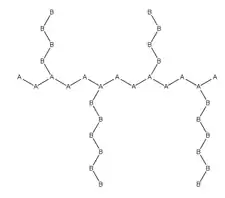
Graft copolymers are a special type of branched copolymer in which the side chains are structurally distinct from the main chain. Typically the main chain is formed from one type of monomer (A) and branches are formed from another monomer (B), or else the side-chains have constitutional or configurational features that differ from those in the main chain.[3]
The individual chains of a graft copolymer may be homopolymers or copolymers. Note that different copolymer sequencing is sufficient to define a structural difference, thus an A-B diblock copolymer with A-B alternating copolymer side chains is properly called a graft copolymer.
For example, polystyrene chains may be grafted onto polybutadiene, a synthetic rubber which retains one reactive C=C double bond per repeat unit. The polybutadiene is dissolved in styrene, which is then subjected to free-radical polymerization. The growing chains can add across the double bonds of rubber molecules forming polystyrene branches. The graft copolymer is formed in a mixture with ungrafted polystyrene chains and rubber molecules.[18]
As with block copolymers, the quasi-composite product has properties of both "components". In the example cited, the rubbery chains absorb energy when the substance is hit, so it is much less brittle than ordinary polystyrene. The product is called high-impact polystyrene, or HIPS.
Star copolymers

Star copolymers have several polymer chains connected to a central core.
Microphase separation

Block copolymers (but not exclusively) are interesting because they can "microphase separate" to form periodic nanostructures,[19][20] as in the styrene-butadiene-styrene block copolymer shown at right. The polymer is known as Kraton and is used for shoe soles and adhesives. Owing to the microfine structure, the transmission electron microscope or TEM was needed to examine the structure. The butadiene matrix was stained with osmium tetroxide to provide contrast in the image. The material was made by living polymerization so that the blocks are almost monodisperse, so helping to create a very regular microstructure. The molecular weight of the polystyrene blocks in the main picture is 102,000; the inset picture has a molecular weight of 91,000, producing slightly smaller domains.
Microphase separation is a situation similar to that of oil and water. Oil and water are immiscible - they phase separate. Due to incompatibility between the blocks, block copolymers undergo a similar phase separation. Because the blocks are covalently bonded to each other, they cannot demix macroscopically as water and oil. In "microphase separation" the blocks form nanometer-sized structures. Depending on the relative lengths of each block, several morphologies can be obtained. In diblock copolymers, sufficiently different block lengths lead to nanometer-sized spheres of one block in a matrix of the second (for example PMMA in polystyrene). Using less different block lengths, a "hexagonally packed cylinder" geometry can be obtained. Blocks of similar length form layers (often called lamellae in the technical literature). Between the cylindrical and lamellar phase is the gyroid phase. The nanoscale structures created from block copolymers could potentially be used for creating devices for use in computer memory, nanoscale-templating and nanoscale separations.[21] Block copolymers are sometimes used as a replacement for phospholipids in model lipid bilayers and liposomes for their superior stability and tunability.[22][23]
Polymer scientists use thermodynamics to describe how the different blocks interact.[24][25] The product of the degree of polymerization, n, and the Flory-Huggins interaction parameter, , gives an indication of how incompatible the two blocks are and whether or not they will microphase separate. For example, a diblock copolymer of symmetric composition will microphase separate if the product is greater than 10.5. If is less than 10.5, the blocks will mix and microphase separation is not observed. The incompatibility between the blocks also affects the solution behavior of these copolymers and their adsorption behavior on various surfaces.[26]


Block (co)polymers are able to self-assemble in selective solvents, to form micelles among other structures.[27]
In thin films, block (co)polymers are of great interest as masks in the lithographic patterning of semiconductor materials for applications in high density data storage. A key challenge is to minimise the feature size and much research is in progress on this.[28]
Copolymer engineering
Copolymerization is used to modify the properties of manufactured plastics to meet specific needs, for example to reduce crystallinity, modify glass transition temperature, control wetting properties or to improve solubility.[29] It is a way of improving mechanical properties, in a technique known as rubber toughening. Elastomeric phases within a rigid matrix act as crack arrestors, and so increase the energy absorption when the material is impacted for example. Acrylonitrile butadiene styrene is a common example.
See also
- Copolymers section of Polymer article
- Thermoplastic elastomer
- Tholin
References
- McNaught, A. D.; Wilkinson, A. (1996). "Glossary of basic terms in polymer science (IUPAC Recommendations 1996)". Pure and Applied Chemistry. 68: 2287–2311. doi:10.1351/goldbook.C01335. ISBN 978-0-9678550-9-7.
- "Nylon-12/6/66 Copolymer". Cosmetics Info. Retrieved 12 April 2021.
- Jenkins, A. D; Kratochvíl, P; Stepto, R. F. T; Suter, U. W (1996). "Glossary of basic terms in polymer science (IUPAC Recommendations 1996)". Pure and Applied Chemistry. 68 (12): 2287–2311. doi:10.1351/pac199668122287.
- Cowie, J.M.G. (1991). Polymers: Chemistry and Physics of Modern Materials (2nd ed.). Blackie (USA: Chapman and Hall). pp. 104–106. ISBN 978-0-216-92980-7.
- Mayo, Frank R.; Lewis, Frederick M. (1944). "Copolymerization. I. A Basis for Comparing the Behavior of Monomers in Copolymerization; The Copolymerization of Styrene and Methyl Methacrylate". J. Am. Chem. Soc. 66 (9): 1594–1601. doi:10.1021/ja01237a052.
- Cowie, p.4
- Hadjichristidis N., Pispas S., Floudas G. Block copolymers: synthetic strategies, physical properties, and applications – Wiley, 2003.
- Fried, Joel R. (2003). Polymer Science and Technology (2nd ed.). Prentice Hall. pp. 41–43. ISBN 978-0-13-018168-8.
- Chum, P. S.; Swogger, K. W. (2008). "Olefin Polymer Technologies-History and Recent Progress at the Dow Chemical Company". Progress in Polymer Science. 33 (8): 797–819. doi:10.1016/j.progpolymsci.2008.05.003.
- Shan, Colin Li Pi; Hazlitt, Lonnie G. (2007). "Block Index for Characterizing Olefin Block Copolymers". Macromol. Symp. 257: 80–93. CiteSeerX 10.1.1.424.4699. doi:10.1002/masy.200751107.
- Painter P. C. and Coleman M. M., Fundamentals of Polymer Science, CRC Press, 1997, p 14.
- Chanda, M. Introduction to Polymer Science and Chemistry. Second Edition. CRC Press, 2013.
- Overberger, C. ″Copolymerization: 1. General Remarks; 2: Selective Examples of Copolymerizations″. Journal of Polymer Science: Polymer Symposium 72, 67-69 (1985).
- Greenley, Robert. ″Free Radical Copolymerization Reactivity Ratios″. The Wiley Database of Polymer Properties. 2003. doi:10.1002/0471532053.bra007
- Ruchatz, Dieter; Fink, Gerhard (1998). "Ethene−Norbornene Copolymerization with Homogeneous Metallocene and Half-Sandwich Catalysts: Kinetics and Relationships between Catalyst Structure and Polymer Structure. 3. Copolymerization Parameters and Copolymerization Diagrams". Macromolecules. 31 (15): 4681–3. Bibcode:1998MaMol..31.4681R. doi:10.1021/ma971043b. PMID 9680398.
- Cao, Ti and Stephen E. Webber. ″Free-Radical Copolymerization of Fullerenes with Styrene″. Macromolecules, 1996, 28, pp 3741-3743.
- Matyjaszewski, Krzysztof (1996). "Controlled radical polymerization". Current Opinion in Solid State and Materials Science. 1 (6): 769–776. Bibcode:1996COSSM...1..769M. doi:10.1016/S1359-0286(96)80101-X.
- Rudin, Alfred (1982). The Elements of Polymer Science and Engineering (1st ed.). Academic Press. p. 19. ISBN 978-0-12-601680-2.
- Hamley, I.W. "The Physics of Block Copolymers" – Oxford University Press, 1998.
- Hamley, I.W. "Developments in Block Copolymer Science and Technology" – Wiley, 2004.
- Gazit, Oz; Khalfin, Rafail; Cohen, Yachin; Tannenbaum, Rina (2009). "Self-assembled diblock copolymer "nanoreactors" as catalysts for metal nanoparticle synthesis". Journal of Physical Chemistry C. 113 (2): 576–583. doi:10.1021/jp807668h.
- Meier, Wolfgang; Nardin, Corinne; Winterhalter, Mathias (2000-12-15). "Reconstitution of Channel Proteins in (Polymerized) ABA Triblock Copolymer Membranes". Angewandte Chemie International Edition. Wiley. 39 (24): 4599–4602. doi:10.1002/1521-3773(20001215)39:24<4599::aid-anie4599>3.0.co;2-y. ISSN 1433-7851. PMID 11169683.
- Zhang, Xiaoyan; Tanner, Pascal; Graff, Alexandra; Palivan, Cornelia G.; Meier, Wolfgang (2012-03-11). "Mimicking the cell membrane with block copolymer membranes". Journal of Polymer Science Part A: Polymer Chemistry. Wiley. 50 (12): 2293–2318. Bibcode:2012JPoSA..50.2293Z. doi:10.1002/pola.26000. ISSN 0887-624X.
- Bates, Frank S.; Fredrickson, Glenn H. (2014). "Block Copolymer Thermodynamics: Theory and Experiment". Annual Review of Physical Chemistry. 41: 525–557. Bibcode:1990ARPC...41..525B. doi:10.1146/annurev.pc.41.100190.002521. PMID 20462355.
- Chremos, Alexandros; Nikoubashman, Arash; Panagiotopoulos, Athanassios (2014). "Flory-Huggins parameter χ, from binary mixtures of Lennard-Jones particles to block copolymer melts". J. Chem. Phys. 140 (5): 054909. Bibcode:2014JChPh.140e4909C. doi:10.1063/1.4863331. PMID 24511981.
- Hershkovitz, Eli; Tannenbaum, Allen; Tannenbaum, Rina (2008). "Adsorption of block co-polymers from selective solvents on curved surfaces". Macromolecules. 41 (9): 3190–3198. Bibcode:2008MaMol..41.3190H. doi:10.1021/ma702706p. PMC 2957843. PMID 20976029.
- Hamley, I.W. "Block Copolymers in Solution" – Wiley, 2005.
- Hamley, IW (2009). "Ordering in Thin Films of Block Copolymers: Fundamentals to Potential Applications". Progress in Polymer Science. 34 (11): 1161–1210. doi:10.1016/j.progpolymsci.2009.06.003.
- Muzammil, Iqbal; Li, Yupeng; Lei, Mingkai (2017). "Tunable wettability and pH-responsiveness of plasma copolymers of acrylic acid and octafluorocyclobutane". Plasma Processes and Polymers. 14 (10): 1700053. doi:10.1002/ppap.201700053. S2CID 104161308.