DNA repair
DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome.[1] In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day.[2] Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages (interstrand crosslinks or ICLs).[3][4] This can eventually lead to malignant tumors, or cancer as per the two hit hypothesis.
The rate of DNA repair is dependent on many factors, including the cell type, the age of the cell, and the extracellular environment. A cell that has accumulated a large amount of DNA damage, or one that no longer effectively repairs damage incurred to its DNA, can enter one of three possible states:
- an irreversible state of dormancy, known as senescence
- cell suicide, also known as apoptosis or programmed cell death
- unregulated cell division, which can lead to the formation of a tumor that is cancerous
The DNA repair ability of a cell is vital to the integrity of its genome and thus to the normal functionality of that organism. Many genes that were initially shown to influence life span have turned out to be involved in DNA damage repair and protection.[5]
The 2015 Nobel Prize in Chemistry was awarded to Tomas Lindahl, Paul Modrich, and Aziz Sancar for their work on the molecular mechanisms of DNA repair processes.[6][7]
DNA damage
DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 10,000 to 1,000,000 molecular lesions per cell per day.[2] While this constitutes only 0.000165% of the human genome's approximately 6 billion bases, unrepaired lesions in critical genes (such as tumor suppressor genes) can impede a cell's ability to carry out its function and appreciably increase the likelihood of tumor formation and contribute to tumour heterogeneity.
The vast majority of DNA damage affects the primary structure of the double helix; that is, the bases themselves are chemically modified. These modifications can in turn disrupt the molecules' regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix. Unlike proteins and RNA, DNA usually lacks tertiary structure and therefore damage or disturbance does not occur at that level. DNA is, however, supercoiled and wound around "packaging" proteins called histones (in eukaryotes), and both superstructures are vulnerable to the effects of DNA damage.
Sources
DNA damage can be subdivided into two main types:
- endogenous damage such as attack by reactive oxygen species produced from normal metabolic byproducts (spontaneous mutation), especially the process of oxidative deamination
- also includes replication errors
- exogenous damage caused by external agents such as
- ultraviolet [UV 200–400 nm] radiation from the sun or other artificial light sources
- other radiation frequencies, including x-rays and gamma rays
- hydrolysis or thermal disruption
- certain plant toxins
- human-made mutagenic chemicals, especially aromatic compounds that act as DNA intercalating agents
- viruses[8]
The replication of damaged DNA before cell division can lead to the incorporation of wrong bases opposite damaged ones. Daughter cells that inherit these wrong bases carry mutations from which the original DNA sequence is unrecoverable (except in the rare case of a back mutation, for example, through gene conversion).
Types
There are several types of damage to DNA due to endogenous cellular processes:
- oxidation of bases [e.g. 8-oxo-7,8-dihydroguanine (8-oxoG)] and generation of DNA strand interruptions from reactive oxygen species,
- alkylation of bases (usually methylation), such as formation of 7-methylguanosine, 1-methyladenine, 6-O-Methylguanine
- hydrolysis of bases, such as deamination, depurination, and depyrimidination.
- "bulky adduct formation" (e.g., benzo[a]pyrene diol epoxide-dG adduct, aristolactam I-dA adduct)
- mismatch of bases, due to errors in DNA replication, in which the wrong DNA base is stitched into place in a newly forming DNA strand, or a DNA base is skipped over or mistakenly inserted.
- Monoadduct damage cause by change in single nitrogenous base of DNA
- Diadduct damage
Damage caused by exogenous agents comes in many forms. Some examples are:
- UV-B light causes crosslinking between adjacent cytosine and thymine bases creating pyrimidine dimers. This is called direct DNA damage.
- UV-A light creates mostly free radicals. The damage caused by free radicals is called indirect DNA damage.
- Ionizing radiation such as that created by radioactive decay or in cosmic rays causes breaks in DNA strands. Intermediate-level ionizing radiation may induce irreparable DNA damage (leading to replicational and transcriptional errors needed for neoplasia or may trigger viral interactions) leading to pre-mature aging and cancer.
- Thermal disruption at elevated temperature increases the rate of depurination (loss of purine bases from the DNA backbone) and single-strand breaks. For example, hydrolytic depurination is seen in the thermophilic bacteria, which grow in hot springs at 40–80 °C.[9][10] The rate of depurination (300 purine residues per genome per generation) is too high in these species to be repaired by normal repair machinery, hence a possibility of an adaptive response cannot be ruled out.
- Industrial chemicals such as vinyl chloride and hydrogen peroxide, and environmental chemicals such as polycyclic aromatic hydrocarbons found in smoke, soot and tar create a huge diversity of DNA adducts- ethenobases, oxidized bases, alkylated phosphotriesters and crosslinking of DNA, just to name a few.
UV damage, alkylation/methylation, X-ray damage and oxidative damage are examples of induced damage. Spontaneous damage can include the loss of a base, deamination, sugar ring puckering and tautomeric shift. Constitutive (spontaneous) DNA damage caused by endogenous oxidants can be detected as a low level of histone H2AX phosphorylation in untreated cells.[11]
Nuclear versus mitochondrial
In human cells, and eukaryotic cells in general, DNA is found in two cellular locations – inside the nucleus and inside the mitochondria. Nuclear DNA (nDNA) exists as chromatin during non-replicative stages of the cell cycle and is condensed into aggregate structures known as chromosomes during cell division. In either state the DNA is highly compacted and wound up around bead-like proteins called histones. Whenever a cell needs to express the genetic information encoded in its nDNA the required chromosomal region is unravelled, genes located therein are expressed, and then the region is condensed back to its resting conformation. Mitochondrial DNA (mtDNA) is located inside mitochondria organelles, exists in multiple copies, and is also tightly associated with a number of proteins to form a complex known as the nucleoid. Inside mitochondria, reactive oxygen species (ROS), or free radicals, byproducts of the constant production of adenosine triphosphate (ATP) via oxidative phosphorylation, create a highly oxidative environment that is known to damage mtDNA. A critical enzyme in counteracting the toxicity of these species is superoxide dismutase, which is present in both the mitochondria and cytoplasm of eukaryotic cells.
Senescence and apoptosis
Senescence, an irreversible process in which the cell no longer divides, is a protective response to the shortening of the chromosome ends, called telomeres. The telomeres are long regions of repetitive noncoding DNA that cap chromosomes and undergo partial degradation each time a cell undergoes division (see Hayflick limit).[12] In contrast, quiescence is a reversible state of cellular dormancy that is unrelated to genome damage (see cell cycle). Senescence in cells may serve as a functional alternative to apoptosis in cases where the physical presence of a cell for spatial reasons is required by the organism,[13] which serves as a "last resort" mechanism to prevent a cell with damaged DNA from replicating inappropriately in the absence of pro-growth cellular signaling. Unregulated cell division can lead to the formation of a tumor (see cancer), which is potentially lethal to an organism. Therefore, the induction of senescence and apoptosis is considered to be part of a strategy of protection against cancer.[14]
Mutation
It is important to distinguish between DNA damage and mutation, the two major types of error in DNA. DNA damage and mutation are fundamentally different. Damage results in physical abnormalities in the DNA, such as single- and double-strand breaks, 8-hydroxydeoxyguanosine residues, and polycyclic aromatic hydrocarbon adducts. DNA damage can be recognized by enzymes, and thus can be correctly repaired if redundant information, such as the undamaged sequence in the complementary DNA strand or in a homologous chromosome, is available for copying. If a cell retains DNA damage, transcription of a gene can be prevented, and thus translation into a protein will also be blocked. Replication may also be blocked or the cell may die.
In contrast to DNA damage, a mutation is a change in the base sequence of the DNA. A mutation cannot be recognized by enzymes once the base change is present in both DNA strands, and thus a mutation cannot be repaired. At the cellular level, mutations can cause alterations in protein function and regulation. Mutations are replicated when the cell replicates. In a population of cells, mutant cells will increase or decrease in frequency according to the effects of the mutation on the ability of the cell to survive and reproduce.
Although distinctly different from each other, DNA damage and mutation are related because DNA damage often causes errors of DNA synthesis during replication or repair; these errors are a major source of mutation.
Given these properties of DNA damage and mutation, it can be seen that DNA damage is a special problem in non-dividing or slowly-dividing cells, where unrepaired damage will tend to accumulate over time. On the other hand, in rapidly dividing cells, unrepaired DNA damage that does not kill the cell by blocking replication will tend to cause replication errors and thus mutation. The great majority of mutations that are not neutral in their effect are deleterious to a cell's survival. Thus, in a population of cells composing a tissue with replicating cells, mutant cells will tend to be lost. However, infrequent mutations that provide a survival advantage will tend to clonally expand at the expense of neighboring cells in the tissue. This advantage to the cell is disadvantageous to the whole organism because such mutant cells can give rise to cancer. Thus, DNA damage in frequently dividing cells, because it gives rise to mutations, is a prominent cause of cancer. In contrast, DNA damage in infrequently-dividing cells is likely a prominent cause of aging.[15]
Mechanisms
Cells cannot function if DNA damage corrupts the integrity and accessibility of essential information in the genome (but cells remain superficially functional when non-essential genes are missing or damaged). Depending on the type of damage inflicted on the DNA's double helical structure, a variety of repair strategies have evolved to restore lost information. If possible, cells use the unmodified complementary strand of the DNA or the sister chromatid as a template to recover the original information. Without access to a template, cells use an error-prone recovery mechanism known as translesion synthesis as a last resort.
Damage to DNA alters the spatial configuration of the helix, and such alterations can be detected by the cell. Once damage is localized, specific DNA repair molecules bind at or near the site of damage, inducing other molecules to bind and form a complex that enables the actual repair to take place.
Direct reversal
Cells are known to eliminate three types of damage to their DNA by chemically reversing it. These mechanisms do not require a template, since the types of damage they counteract can occur in only one of the four bases. Such direct reversal mechanisms are specific to the type of damage incurred and do not involve breakage of the phosphodiester backbone. The formation of pyrimidine dimers upon irradiation with UV light results in an abnormal covalent bond between adjacent pyrimidine bases. The photoreactivation process directly reverses this damage by the action of the enzyme photolyase, whose activation is obligately dependent on energy absorbed from blue/UV light (300–500 nm wavelength) to promote catalysis.[16] Photolyase, an old enzyme present in bacteria, fungi, and most animals no longer functions in humans,[17] who instead use nucleotide excision repair to repair damage from UV irradiation. Another type of damage, methylation of guanine bases, is directly reversed by the protein methyl guanine methyl transferase (MGMT), the bacterial equivalent of which is called ogt. This is an expensive process because each MGMT molecule can be used only once; that is, the reaction is stoichiometric rather than catalytic.[18] A generalized response to methylating agents in bacteria is known as the adaptive response and confers a level of resistance to alkylating agents upon sustained exposure by upregulation of alkylation repair enzymes.[19] The third type of DNA damage reversed by cells is certain methylation of the bases cytosine and adenine.
Single-strand damage
When only one of the two strands of a double helix has a defect, the other strand can be used as a template to guide the correction of the damaged strand. In order to repair damage to one of the two paired molecules of DNA, there exist a number of excision repair mechanisms that remove the damaged nucleotide and replace it with an undamaged nucleotide complementary to that found in the undamaged DNA strand.[18]
- Base excision repair (BER): damaged single bases or nucleotides are most commonly repaired by removing the base or the nucleotide involved and then inserting the correct base or nucleotide. In base excision repair, a glycosylase[20] enzyme removes the damaged base from the DNA by cleaving the bond between the base and the deoxyribose. These enzymes remove a single base to create an apurinic or apyrimidinic site (AP site).[20] Enzymes called AP endonucleases nick the damaged DNA backbone at the AP site. DNA polymerase then removes the damaged region using its 5’ to 3’ exonuclease activity and correctly synthesizes the new strand using the complementary strand as a template.[20] The gap is then sealed by enzyme DNA ligase.[21]
- Nucleotide excision repair (NER): bulky, helix-distorting damage, such as pyrimidine dimerization caused by UV light is usually repaired by a three-step process. First the damage is recognized, then 12-24 nucleotide-long strands of DNA are removed both upstream and downstream of the damage site by endonucleases, and the removed DNA region is then resynthesized.[22] NER is a highly evolutionarily conserved repair mechanism and is used in nearly all eukaryotic and prokaryotic cells.[22] In prokaryotes, NER is mediated by Uvr proteins.[22] In eukaryotes, many more proteins are involved, although the general strategy is the same.[22]
- Mismatch repair systems are present in essentially all cells to correct errors that are not corrected by proofreading. These systems consist of at least two proteins. One detects the mismatch, and the other recruits an endonuclease that cleaves the newly synthesized DNA strand close to the region of damage. In E. coli , the proteins involved are the Mut class proteins: MutS, MutL, and MutH. In most Eukaryotes, the analog for MutS is MSH and the analog for MutL is MLH. MutH is only present in bacteria. This is followed by removal of damaged region by an exonuclease, resynthesis by DNA polymerase, and nick sealing by DNA ligase.[23]
Double-strand breaks
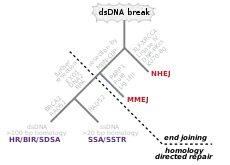
Double-strand breaks, in which both strands in the double helix are severed, are particularly hazardous to the cell because they can lead to genome rearrangements. In fact, when a double-strand break is accompanied by a cross-linkage joining the two strands at the same point, neither strand can be used as a template for the repair mechanisms, so that the cell will not be able to complete mitosis when it next divides, and will either die or, in rare cases, undergo a mutation.[3][4] Three mechanisms exist to repair double-strand breaks (DSBs): non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination (HR):[18][24]
- In NHEJ, DNA Ligase IV, a specialized DNA ligase that forms a complex with the cofactor XRCC4, directly joins the two ends.[25] To guide accurate repair, NHEJ relies on short homologous sequences called microhomologies present on the single-stranded tails of the DNA ends to be joined. If these overhangs are compatible, repair is usually accurate.[26][27][28][29] NHEJ can also introduce mutations during repair. Loss of damaged nucleotides at the break site can lead to deletions, and joining of nonmatching termini forms insertions or translocations. NHEJ is especially important before the cell has replicated its DNA, since there is no template available for repair by homologous recombination. There are "backup" NHEJ pathways in higher eukaryotes.[30] Besides its role as a genome caretaker, NHEJ is required for joining hairpin-capped double-strand breaks induced during V(D)J recombination, the process that generates diversity in B-cell and T-cell receptors in the vertebrate immune system.[31]
- MMEJ starts with short-range end resection by MRE11 nuclease on either side of a double-strand break to reveal microhomology regions.[32] In further steps,[33] Poly (ADP-ribose) polymerase 1 (PARP1) is required and may be an early step in MMEJ. There is pairing of microhomology regions followed by recruitment of flap structure-specific endonuclease 1 (FEN1) to remove overhanging flaps. This is followed by recruitment of XRCC1–LIG3 to the site for ligating the DNA ends, leading to an intact DNA. MMEJ is always accompanied by a deletion, so that MMEJ is a mutagenic pathway for DNA repair.[34]
- HR requires the presence of an identical or nearly identical sequence to be used as a template for repair of the break. The enzymatic machinery responsible for this repair process is nearly identical to the machinery responsible for chromosomal crossover during meiosis. This pathway allows a damaged chromosome to be repaired using a sister chromatid (available in G2 after DNA replication) or a homologous chromosome as a template. DSBs caused by the replication machinery attempting to synthesize across a single-strand break or unrepaired lesion cause collapse of the replication fork and are typically repaired by recombination.
In an in vitro system, MMEJ occurred in mammalian cells at the levels of 10–20% of HR when both HR and NHEJ mechanisms were also available.[32]
The extremophile Deinococcus radiodurans has a remarkable ability to survive DNA damage from ionizing radiation and other sources. At least two copies of the genome, with random DNA breaks, can form DNA fragments through annealing. Partially overlapping fragments are then used for synthesis of homologous regions through a moving D-loop that can continue extension until complementary partner strands are found. In the final step, there is crossover by means of RecA-dependent homologous recombination.[35]
Topoisomerases introduce both single- and double-strand breaks in the course of changing the DNA's state of supercoiling, which is especially common in regions near an open replication fork. Such breaks are not considered DNA damage because they are a natural intermediate in the topoisomerase biochemical mechanism and are immediately repaired by the enzymes that created them.
Another type of DNA double-strand breaks originates from the DNA heat-sensitive or heat-labile sites. These DNA sites are not initial DSBs. However, they convert to DSB after treating with elevated temperature. Ionizing irradiation can induces a highly complex form of DNA damage as clustered damage. It consists of different types of DNA lesions in various locations of the DNA helix. Some of these closely located lesions can probably convert to DSB by exposure to high temperatures. But the exact nature of these lesions and their interactions is not yet known[36]
Translesion synthesis
Translesion synthesis (TLS) is a DNA damage tolerance process that allows the DNA replication machinery to replicate past DNA lesions such as thymine dimers or AP sites.[37] It involves switching out regular DNA polymerases for specialized translesion polymerases (i.e. DNA polymerase IV or V, from the Y Polymerase family), often with larger active sites that can facilitate the insertion of bases opposite damaged nucleotides. The polymerase switching is thought to be mediated by, among other factors, the post-translational modification of the replication processivity factor PCNA. Translesion synthesis polymerases often have low fidelity (high propensity to insert wrong bases) on undamaged templates relative to regular polymerases. However, many are extremely efficient at inserting correct bases opposite specific types of damage. For example, Pol η mediates error-free bypass of lesions induced by UV irradiation, whereas Pol ι introduces mutations at these sites. Pol η is known to add the first adenine across the T^T photodimer using Watson-Crick base pairing and the second adenine will be added in its syn conformation using Hoogsteen base pairing. From a cellular perspective, risking the introduction of point mutations during translesion synthesis may be preferable to resorting to more drastic mechanisms of DNA repair, which may cause gross chromosomal aberrations or cell death. In short, the process involves specialized polymerases either bypassing or repairing lesions at locations of stalled DNA replication. For example, Human DNA polymerase eta can bypass complex DNA lesions like guanine-thymine intra-strand crosslink, G[8,5-Me]T, although it can cause targeted and semi-targeted mutations.[38] Paromita Raychaudhury and Ashis Basu[39] studied the toxicity and mutagenesis of the same lesion in Escherichia coli by replicating a G[8,5-Me]T-modified plasmid in E. coli with specific DNA polymerase knockouts. Viability was very low in a strain lacking pol II, pol IV, and pol V, the three SOS-inducible DNA polymerases, indicating that translesion synthesis is conducted primarily by these specialized DNA polymerases. A bypass platform is provided to these polymerases by Proliferating cell nuclear antigen (PCNA). Under normal circumstances, PCNA bound to polymerases replicates the DNA. At a site of lesion, PCNA is ubiquitinated, or modified, by the RAD6/RAD18 proteins to provide a platform for the specialized polymerases to bypass the lesion and resume DNA replication.[40][41] After translesion synthesis, extension is required. This extension can be carried out by a replicative polymerase if the TLS is error-free, as in the case of Pol η, yet if TLS results in a mismatch, a specialized polymerase is needed to extend it; Pol ζ. Pol ζ is unique in that it can extend terminal mismatches, whereas more processive polymerases cannot. So when a lesion is encountered, the replication fork will stall, PCNA will switch from a processive polymerase to a TLS polymerase such as Pol ι to fix the lesion, then PCNA may switch to Pol ζ to extend the mismatch, and last PCNA will switch to the processive polymerase to continue replication.
Global response to DNA damage
Cells exposed to ionizing radiation, ultraviolet light or chemicals are prone to acquire multiple sites of bulky DNA lesions and double-strand breaks. Moreover, DNA damaging agents can damage other biomolecules such as proteins, carbohydrates, lipids, and RNA. The accumulation of damage, to be specific, double-strand breaks or adducts stalling the replication forks, are among known stimulation signals for a global response to DNA damage.[42] The global response to damage is an act directed toward the cells' own preservation and triggers multiple pathways of macromolecular repair, lesion bypass, tolerance, or apoptosis. The common features of global response are induction of multiple genes, cell cycle arrest, and inhibition of cell division.
Initial steps
The packaging of eukaryotic DNA into chromatin presents a barrier to all DNA-based processes that require recruitment of enzymes to their sites of action. To allow DNA repair, the chromatin must be remodeled. In eukaryotes, ATP dependent chromatin remodeling complexes and histone-modifying enzymes are two predominant factors employed to accomplish this remodeling process.[43]
Chromatin relaxation occurs rapidly at the site of a DNA damage.[44][45] In one of the earliest steps, the stress-activated protein kinase, c-Jun N-terminal kinase (JNK), phosphorylates SIRT6 on serine 10 in response to double-strand breaks or other DNA damage.[46] This post-translational modification facilitates the mobilization of SIRT6 to DNA damage sites, and is required for efficient recruitment of poly (ADP-ribose) polymerase 1 (PARP1) to DNA break sites and for efficient repair of DSBs.[46] PARP1 protein starts to appear at DNA damage sites in less than a second, with half maximum accumulation within 1.6 seconds after the damage occurs.[47] PARP1 synthesizes polymeric adenosine diphosphate ribose (poly (ADP-ribose) or PAR) chains on itself. Next the chromatin remodeler ALC1 quickly attaches to the product of PARP1 action, a poly-ADP ribose chain, and ALC1 completes arrival at the DNA damage within 10 seconds of the occurrence of the damage.[45] About half of the maximum chromatin relaxation, presumably due to action of ALC1, occurs by 10 seconds.[45] This then allows recruitment of the DNA repair enzyme MRE11, to initiate DNA repair, within 13 seconds.[47]
γH2AX, the phosphorylated form of H2AX is also involved in the early steps leading to chromatin decondensation after DNA double-strand breaks. The histone variant H2AX constitutes about 10% of the H2A histones in human chromatin.[48] γH2AX (H2AX phosphorylated on serine 139) can be detected as soon as 20 seconds after irradiation of cells (with DNA double-strand break formation), and half maximum accumulation of γH2AX occurs in one minute.[48] The extent of chromatin with phosphorylated γH2AX is about two million base pairs at the site of a DNA double-strand break.[48] γH2AX does not, itself, cause chromatin decondensation, but within 30 seconds of irradiation, RNF8 protein can be detected in association with γH2AX.[49] RNF8 mediates extensive chromatin decondensation, through its subsequent interaction with CHD4,[50] a component of the nucleosome remodeling and deacetylase complex NuRD.
DDB2 occurs in a heterodimeric complex with DDB1. This complex further complexes with the ubiquitin ligase protein CUL4A[51] and with PARP1.[52] This larger complex rapidly associates with UV-induced damage within chromatin, with half-maximum association completed in 40 seconds.[51] The PARP1 protein, attached to both DDB1 and DDB2, then PARylates (creates a poly-ADP ribose chain) on DDB2 that attracts the DNA remodeling protein ALC1.[52] Action of ALC1 relaxes the chromatin at the site of UV damage to DNA. This relaxation allows other proteins in the nucleotide excision repair pathway to enter the chromatin and repair UV-induced cyclobutane pyrimidine dimer damages.
After rapid chromatin remodeling, cell cycle checkpoints are activated to allow DNA repair to occur before the cell cycle progresses. First, two kinases, ATM and ATR are activated within 5 or 6 minutes after DNA is damaged. This is followed by phosphorylation of the cell cycle checkpoint protein Chk1, initiating its function, about 10 minutes after DNA is damaged.[53]
DNA damage checkpoints
After DNA damage, cell cycle checkpoints are activated. Checkpoint activation pauses the cell cycle and gives the cell time to repair the damage before continuing to divide. DNA damage checkpoints occur at the G1/S and G2/M boundaries. An intra-S checkpoint also exists. Checkpoint activation is controlled by two master kinases, ATM and ATR. ATM responds to DNA double-strand breaks and disruptions in chromatin structure,[54] whereas ATR primarily responds to stalled replication forks. These kinases phosphorylate downstream targets in a signal transduction cascade, eventually leading to cell cycle arrest. A class of checkpoint mediator proteins including BRCA1, MDC1, and 53BP1 has also been identified.[55] These proteins seem to be required for transmitting the checkpoint activation signal to downstream proteins.
DNA damage checkpoint is a signal transduction pathway that blocks cell cycle progression in G1, G2 and metaphase and slows down the rate of S phase progression when DNA is damaged. It leads to a pause in cell cycle allowing the cell time to repair the damage before continuing to divide.
Checkpoint Proteins can be separated into four groups: phosphatidylinositol 3-kinase (PI3K)-like protein kinase, proliferating cell nuclear antigen (PCNA)-like group, two serine/threonine(S/T) kinases and their adaptors. Central to all DNA damage induced checkpoints responses is a pair of large protein kinases belonging to the first group of PI3K-like protein kinases-the ATM (Ataxia telangiectasia mutated) and ATR (Ataxia- and Rad-related) kinases, whose sequence and functions have been well conserved in evolution. All DNA damage response requires either ATM or ATR because they have the ability to bind to the chromosomes at the site of DNA damage, together with accessory proteins that are platforms on which DNA damage response components and DNA repair complexes can be assembled.
An important downstream target of ATM and ATR is p53, as it is required for inducing apoptosis following DNA damage.[56] The cyclin-dependent kinase inhibitor p21 is induced by both p53-dependent and p53-independent mechanisms and can arrest the cell cycle at the G1/S and G2/M checkpoints by deactivating cyclin/cyclin-dependent kinase complexes.[57]
The prokaryotic SOS response
The SOS response is the changes in gene expression in Escherichia coli and other bacteria in response to extensive DNA damage. The prokaryotic SOS system is regulated by two key proteins: LexA and RecA. The LexA homodimer is a transcriptional repressor that binds to operator sequences commonly referred to as SOS boxes. In Escherichia coli it is known that LexA regulates transcription of approximately 48 genes including the lexA and recA genes.[58] The SOS response is known to be widespread in the Bacteria domain, but it is mostly absent in some bacterial phyla, like the Spirochetes.[59] The most common cellular signals activating the SOS response are regions of single-stranded DNA (ssDNA), arising from stalled replication forks or double-strand breaks, which are processed by DNA helicase to separate the two DNA strands.[42] In the initiation step, RecA protein binds to ssDNA in an ATP hydrolysis driven reaction creating RecA–ssDNA filaments. RecA–ssDNA filaments activate LexA autoprotease activity, which ultimately leads to cleavage of LexA dimer and subsequent LexA degradation. The loss of LexA repressor induces transcription of the SOS genes and allows for further signal induction, inhibition of cell division and an increase in levels of proteins responsible for damage processing.
In Escherichia coli, SOS boxes are 20-nucleotide long sequences near promoters with palindromic structure and a high degree of sequence conservation. In other classes and phyla, the sequence of SOS boxes varies considerably, with different length and composition, but it is always highly conserved and one of the strongest short signals in the genome.[59] The high information content of SOS boxes permits differential binding of LexA to different promoters and allows for timing of the SOS response. The lesion repair genes are induced at the beginning of SOS response. The error-prone translesion polymerases, for example, UmuCD'2 (also called DNA polymerase V), are induced later on as a last resort.[60] Once the DNA damage is repaired or bypassed using polymerases or through recombination, the amount of single-stranded DNA in cells is decreased, lowering the amounts of RecA filaments decreases cleavage activity of LexA homodimer, which then binds to the SOS boxes near promoters and restores normal gene expression.
Eukaryotic transcriptional responses to DNA damage
Eukaryotic cells exposed to DNA damaging agents also activate important defensive pathways by inducing multiple proteins involved in DNA repair, cell cycle checkpoint control, protein trafficking and degradation. Such genome wide transcriptional response is very complex and tightly regulated, thus allowing coordinated global response to damage. Exposure of yeast Saccharomyces cerevisiae to DNA damaging agents results in overlapping but distinct transcriptional profiles. Similarities to environmental shock response indicates that a general global stress response pathway exist at the level of transcriptional activation. In contrast, different human cell types respond to damage differently indicating an absence of a common global response. The probable explanation for this difference between yeast and human cells may be in the heterogeneity of mammalian cells. In an animal different types of cells are distributed among different organs that have evolved different sensitivities to DNA damage.[61]
In general global response to DNA damage involves expression of multiple genes responsible for postreplication repair, homologous recombination, nucleotide excision repair, DNA damage checkpoint, global transcriptional activation, genes controlling mRNA decay, and many others. A large amount of damage to a cell leaves it with an important decision: undergo apoptosis and die, or survive at the cost of living with a modified genome. An increase in tolerance to damage can lead to an increased rate of survival that will allow a greater accumulation of mutations. Yeast Rev1 and human polymerase η are members of Y family translesion DNA polymerases present during global response to DNA damage and are responsible for enhanced mutagenesis during a global response to DNA damage in eukaryotes.[42]
Aging
Pathological effects of poor DNA repair
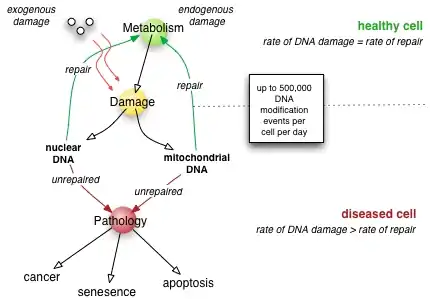
Experimental animals with genetic deficiencies in DNA repair often show decreased life span and increased cancer incidence.[15] For example, mice deficient in the dominant NHEJ pathway and in telomere maintenance mechanisms get lymphoma and infections more often, and, as a consequence, have shorter lifespans than wild-type mice.[62] In similar manner, mice deficient in a key repair and transcription protein that unwinds DNA helices have premature onset of aging-related diseases and consequent shortening of lifespan.[63] However, not every DNA repair deficiency creates exactly the predicted effects; mice deficient in the NER pathway exhibited shortened life span without correspondingly higher rates of mutation.[64]
If the rate of DNA damage exceeds the capacity of the cell to repair it, the accumulation of errors can overwhelm the cell and result in early senescence, apoptosis, or cancer. Inherited diseases associated with faulty DNA repair functioning result in premature aging,[15] increased sensitivity to carcinogens and correspondingly increased cancer risk (see below). On the other hand, organisms with enhanced DNA repair systems, such as Deinococcus radiodurans, the most radiation-resistant known organism, exhibit remarkable resistance to the double-strand break-inducing effects of radioactivity, likely due to enhanced efficiency of DNA repair and especially NHEJ.[65]
Longevity and caloric restriction
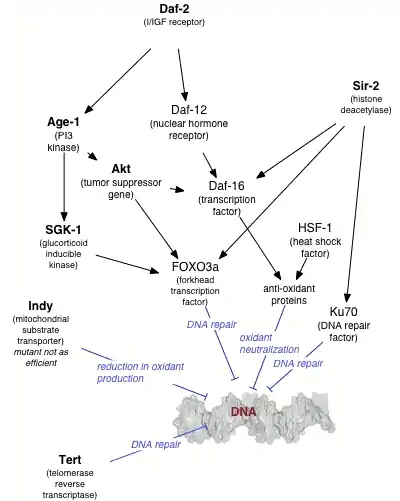
A number of individual genes have been identified as influencing variations in life span within a population of organisms. The effects of these genes is strongly dependent on the environment, in particular, on the organism's diet. Caloric restriction reproducibly results in extended lifespan in a variety of organisms, likely via nutrient sensing pathways and decreased metabolic rate. The molecular mechanisms by which such restriction results in lengthened lifespan are as yet unclear (see[66] for some discussion); however, the behavior of many genes known to be involved in DNA repair is altered under conditions of caloric restriction. Several agents reported to have anti-aging properties have been shown to attenuate constitutive level of mTOR signaling, an evidence of reduction of metabolic activity, and concurrently to reduce constitutive level of DNA damage induced by endogenously generated reactive oxygen species.[67]
For example, increasing the gene dosage of the gene SIR-2, which regulates DNA packaging in the nematode worm Caenorhabditis elegans, can significantly extend lifespan.[68] The mammalian homolog of SIR-2 is known to induce downstream DNA repair factors involved in NHEJ, an activity that is especially promoted under conditions of caloric restriction.[69] Caloric restriction has been closely linked to the rate of base excision repair in the nuclear DNA of rodents,[70] although similar effects have not been observed in mitochondrial DNA.[71]
The C. elegans gene AGE-1, an upstream effector of DNA repair pathways, confers dramatically extended life span under free-feeding conditions but leads to a decrease in reproductive fitness under conditions of caloric restriction.[72] This observation supports the pleiotropy theory of the biological origins of aging, which suggests that genes conferring a large survival advantage early in life will be selected for even if they carry a corresponding disadvantage late in life.
Medicine and DNA repair modulation
Hereditary DNA repair disorders
Defects in the NER mechanism are responsible for several genetic disorders, including:
- Xeroderma pigmentosum: hypersensitivity to sunlight/UV, resulting in increased skin cancer incidence and premature aging
- Cockayne syndrome: hypersensitivity to UV and chemical agents
- Trichothiodystrophy: sensitive skin, brittle hair and nails
Mental retardation often accompanies the latter two disorders, suggesting increased vulnerability of developmental neurons.
Other DNA repair disorders include:
- Werner's syndrome: premature aging and retarded growth
- Bloom's syndrome: sunlight hypersensitivity, high incidence of malignancies (especially leukemias).
- Ataxia telangiectasia: sensitivity to ionizing radiation and some chemical agents
All of the above diseases are often called "segmental progerias" ("accelerated aging diseases") because those affected appear elderly and experience aging-related diseases at an abnormally young age, while not manifesting all the symptoms of old age.
Other diseases associated with reduced DNA repair function include Fanconi anemia, hereditary breast cancer and hereditary colon cancer.
Cancer
Because of inherent limitations in the DNA repair mechanisms, if humans lived long enough, they would all eventually develop cancer.[73][74] There are at least 34 Inherited human DNA repair gene mutations that increase cancer risk. Many of these mutations cause DNA repair to be less effective than normal. In particular, Hereditary nonpolyposis colorectal cancer (HNPCC) is strongly associated with specific mutations in the DNA mismatch repair pathway. BRCA1 and BRCA2, two important genes whose mutations confer a hugely increased risk of breast cancer on carriers,[75] are both associated with a large number of DNA repair pathways, especially NHEJ and homologous recombination.
Cancer therapy procedures such as chemotherapy and radiotherapy work by overwhelming the capacity of the cell to repair DNA damage, resulting in cell death. Cells that are most rapidly dividing – most typically cancer cells – are preferentially affected. The side-effect is that other non-cancerous but rapidly dividing cells such as progenitor cells in the gut, skin, and hematopoietic system are also affected. Modern cancer treatments attempt to localize the DNA damage to cells and tissues only associated with cancer, either by physical means (concentrating the therapeutic agent in the region of the tumor) or by biochemical means (exploiting a feature unique to cancer cells in the body). In the context of therapies targeting DNA damage response genes, the latter approach has been termed 'synthetic lethality'.[76]
Perhaps the most well-known of these 'synthetic lethality' drugs is the poly(ADP-ribose) polymerase 1 (PARP1) inhibitor olaparib, which was approved by the Food and Drug Administration in 2015 for the treatment in women of BRCA-defective ovarian cancer. Tumor cells with partial loss of DNA damage response (specifically, homologous recombination repair) are dependent on another mechanism – single-strand break repair – which is a mechanism consisting, in part, of the PARP1 gene product.[77] Olaparib is combined with chemotherapeutics to inhibit single-strand break repair induced by DNA damage caused by the co-administered chemotherapy. Tumor cells relying on this residual DNA repair mechanism are unable to repair the damage and hence are not able to survive and proliferate, whereas normal cells can repair the damage with the functioning homologous recombination mechanism.
Many other drugs for use against other residual DNA repair mechanisms commonly found in cancer are currently under investigation. However, synthetic lethality therapeutic approaches have been questioned due to emerging evidence of acquired resistance, achieved through rewiring of DNA damage response pathways and reversion of previously inhibited defects.[78]
DNA repair defects in cancer
It has become apparent over the past several years that the DNA damage response acts as a barrier to the malignant transformation of preneoplastic cells.[79] Previous studies have shown an elevated DNA damage response in cell-culture models with oncogene activation[80] and preneoplastic colon adenomas.[81] DNA damage response mechanisms trigger cell-cycle arrest, and attempt to repair DNA lesions or promote cell death/senescence if repair is not possible. Replication stress is observed in preneoplastic cells due to increased proliferation signals from oncogenic mutations. Replication stress is characterized by: increased replication initiation/origin firing; increased transcription and collisions of transcription-replication complexes; nucleotide deficiency; increase in reactive oxygen species (ROS).[82]
Replication stress, along with the selection for inactivating mutations in DNA damage response genes in the evolution of the tumor,[83] leads to downregulation and/or loss of some DNA damage response mechanisms, and hence loss of DNA repair and/or senescence/programmed cell death. In experimental mouse models, loss of DNA damage response-mediated cell senescence was observed after using a short hairpin RNA (shRNA) to inhibit the double-strand break response kinase ataxia telangiectasia (ATM), leading to increased tumor size and invasiveness.[81] Humans born with inherited defects in DNA repair mechanisms (for example, Li-Fraumeni syndrome) have a higher cancer risk.[84]
The prevalence of DNA damage response mutations differs across cancer types; for example, 30% of breast invasive carcinomas have mutations in genes involved in homologous recombination.[79] In cancer, downregulation is observed across all DNA damage response mechanisms (base excision repair (BER), nucleotide excision repair (NER), DNA mismatch repair (MMR), homologous recombination repair (HR), non-homologous end joining (NHEJ) and translesion DNA synthesis (TLS).[85] As well as mutations to DNA damage repair genes, mutations also arise in the genes responsible for arresting the cell cycle to allow sufficient time for DNA repair to occur, and some genes are involved in both DNA damage repair and cell cycle checkpoint control, for example ATM and checkpoint kinase 2 (CHEK2) – a tumor suppressor that is often absent or downregulated in non-small cell lung cancer.[86]
| HR | NHEJ | SSA | FA | BER | NER | MMR | |
|---|---|---|---|---|---|---|---|
| ATM | |||||||
| ATR | |||||||
| PAXIP | |||||||
| RPA | |||||||
| BRCA1 | |||||||
| BRCA2 | |||||||
| RAD51 | |||||||
| RFC | |||||||
| XRCC1 | |||||||
| PCNA | |||||||
| PARP1 | |||||||
| ERCC1 | |||||||
| MSH3 |
Epigenetic DNA repair defects in cancer
Classically, cancer has been viewed as a set of diseases that are driven by progressive genetic abnormalities that include mutations in tumour-suppressor genes and oncogenes, and chromosomal aberrations. However, it has become apparent that cancer is also driven by epigenetic alterations.[87]
Epigenetic alterations refer to functionally relevant modifications to the genome that do not involve a change in the nucleotide sequence. Examples of such modifications are changes in DNA methylation (hypermethylation and hypomethylation) and histone modification,[88] changes in chromosomal architecture (caused by inappropriate expression of proteins such as HMGA2 or HMGA1)[89] and changes caused by microRNAs. Each of these epigenetic alterations serves to regulate gene expression without altering the underlying DNA sequence. These changes usually remain through cell divisions, last for multiple cell generations, and can be considered to be epimutations (equivalent to mutations).
While large numbers of epigenetic alterations are found in cancers, the epigenetic alterations in DNA repair genes, causing reduced expression of DNA repair proteins, appear to be particularly important. Such alterations are thought to occur early in progression to cancer and to be a likely cause of the genetic instability characteristic of cancers.[90][91][92]
Reduced expression of DNA repair genes causes deficient DNA repair. When DNA repair is deficient DNA damages remain in cells at a higher than usual level and these excess damages cause increased frequencies of mutation or epimutation. Mutation rates increase substantially in cells defective in DNA mismatch repair[93][94] or in homologous recombinational repair (HRR).[95] Chromosomal rearrangements and aneuploidy also increase in HRR defective cells.[96]
Higher levels of DNA damage not only cause increased mutation, but also cause increased epimutation. During repair of DNA double strand breaks, or repair of other DNA damages, incompletely cleared sites of repair can cause epigenetic gene silencing.[97][98]
Deficient expression of DNA repair proteins due to an inherited mutation can cause increased risk of cancer. Individuals with an inherited impairment in any of 34 DNA repair genes (see article DNA repair-deficiency disorder) have an increased risk of cancer, with some defects causing up to a 100% lifetime chance of cancer (e.g. p53 mutations).[99] However, such germline mutations (which cause highly penetrant cancer syndromes) are the cause of only about 1 percent of cancers.[100]
Frequencies of epimutations in DNA repair genes
Deficiencies in DNA repair enzymes are occasionally caused by a newly arising somatic mutation in a DNA repair gene, but are much more frequently caused by epigenetic alterations that reduce or silence expression of DNA repair genes. For example, when 113 colorectal cancers were examined in sequence, only four had a missense mutation in the DNA repair gene MGMT, while the majority had reduced MGMT expression due to methylation of the MGMT promoter region (an epigenetic alteration).[101] Five different studies found that between 40% and 90% of colorectal cancers have reduced MGMT expression due to methylation of the MGMT promoter region.[102][103][104][105][106]
Similarly, out of 119 cases of mismatch repair-deficient colorectal cancers that lacked DNA repair gene PMS2 expression, PMS2 was deficient in 6 due to mutations in the PMS2 gene, while in 103 cases PMS2 expression was deficient because its pairing partner MLH1 was repressed due to promoter methylation (PMS2 protein is unstable in the absence of MLH1).[107] In the other 10 cases, loss of PMS2 expression was likely due to epigenetic overexpression of the microRNA, miR-155, which down-regulates MLH1.[108]
In a further example, epigenetic defects were found in various cancers (e.g. breast, ovarian, colorectal and head and neck). Two or three deficiencies in the expression of ERCC1, XPF or PMS2 occur simultaneously in the majority of 49 colon cancers evaluated by Facista et al.[109]
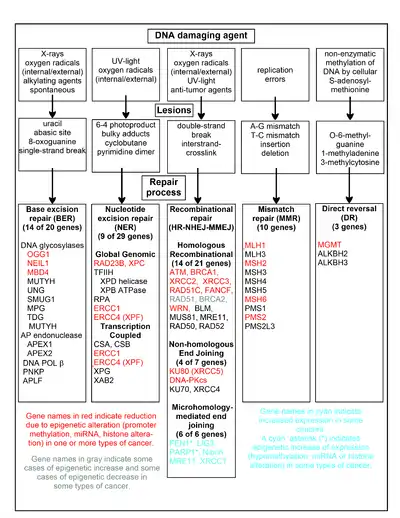
The chart in this section shows some frequent DNA damaging agents, examples of DNA lesions they cause, and the pathways that deal with these DNA damages. At least 169 enzymes are either directly employed in DNA repair or influence DNA repair processes.[110] Of these, 83 are directly employed in repairing the 5 types of DNA damages illustrated in the chart.
Some of the more well studied genes central to these repair processes are shown in the chart. The gene designations shown in red, gray or cyan indicate genes frequently epigenetically altered in various types of cancers. Wikipedia articles on each of the genes highlighted by red, gray or cyan describe the epigenetic alteration(s) and the cancer(s) in which these epimutations are found. Review articles,[111] and broad experimental survey articles[112][113] also document most of these epigenetic DNA repair deficiencies in cancers.
Red-highlighted genes are frequently reduced or silenced by epigenetic mechanisms in various cancers. When these genes have low or absent expression, DNA damages can accumulate. Replication errors past these damages (see translesion synthesis) can lead to increased mutations and, ultimately, cancer. Epigenetic repression of DNA repair genes in accurate DNA repair pathways appear to be central to carcinogenesis.
The two gray-highlighted genes RAD51 and BRCA2, are required for homologous recombinational repair. They are sometimes epigenetically over-expressed and sometimes under-expressed in certain cancers. As indicated in the Wikipedia articles on RAD51 and BRCA2, such cancers ordinarily have epigenetic deficiencies in other DNA repair genes. These repair deficiencies would likely cause increased unrepaired DNA damages. The over-expression of RAD51 and BRCA2 seen in these cancers may reflect selective pressures for compensatory RAD51 or BRCA2 over-expression and increased homologous recombinational repair to at least partially deal with such excess DNA damages. In those cases where RAD51 or BRCA2 are under-expressed, this would itself lead to increased unrepaired DNA damages. Replication errors past these damages (see translesion synthesis) could cause increased mutations and cancer, so that under-expression of RAD51 or BRCA2 would be carcinogenic in itself.
Cyan-highlighted genes are in the microhomology-mediated end joining (MMEJ) pathway and are up-regulated in cancer. MMEJ is an additional error-prone inaccurate repair pathway for double-strand breaks. In MMEJ repair of a double-strand break, an homology of 5–25 complementary base pairs between both paired strands is sufficient to align the strands, but mismatched ends (flaps) are usually present. MMEJ removes the extra nucleotides (flaps) where strands are joined, and then ligates the strands to create an intact DNA double helix. MMEJ almost always involves at least a small deletion, so that it is a mutagenic pathway.[24] FEN1, the flap endonuclease in MMEJ, is epigenetically increased by promoter hypomethylation and is over-expressed in the majority of cancers of the breast,[114] prostate,[115] stomach,[116][117] neuroblastomas,[118] pancreas,[119] and lung.[120] PARP1 is also over-expressed when its promoter region ETS site is epigenetically hypomethylated, and this contributes to progression to endometrial cancer[121] and BRCA-mutated serous ovarian cancer.[122] Other genes in the MMEJ pathway are also over-expressed in a number of cancers (see MMEJ for summary), and are also shown in cyan.
Genome-wide distribution of DNA repair in human somatic cells
Differential activity of DNA repair pathways across various regions of the human genome causes mutations to be very unevenly distributed within tumor genomes.[123][124] In particular, the gene-rich, early-replicating regions of the human genome exhibit lower mutation frequencies than the gene-poor, late-replicating heterochromatin. One mechanism underlying this involves the histone modification H3K36me3, which can recruit mismatch repair proteins,[125] thereby lowering mutation rates in H3K36me3-marked regions.[126] Another important mechanism concerns nucleotide excision repair, which can be recruited by the transcription machinery, lowering somatic mutation rates in active genes[124] and other open chromatin regions.[127]
Evolution
The basic processes of DNA repair are highly conserved among both prokaryotes and eukaryotes and even among bacteriophages (viruses which infect bacteria); however, more complex organisms with more complex genomes have correspondingly more complex repair mechanisms.[128] The ability of a large number of protein structural motifs to catalyze relevant chemical reactions has played a significant role in the elaboration of repair mechanisms during evolution. For an extremely detailed review of hypotheses relating to the evolution of DNA repair, see.[129]
The fossil record indicates that single-cell life began to proliferate on the planet at some point during the Precambrian period, although exactly when recognizably modern life first emerged is unclear. Nucleic acids became the sole and universal means of encoding genetic information, requiring DNA repair mechanisms that in their basic form have been inherited by all extant life forms from their common ancestor. The emergence of Earth's oxygen-rich atmosphere (known as the "oxygen catastrophe") due to photosynthetic organisms, as well as the presence of potentially damaging free radicals in the cell due to oxidative phosphorylation, necessitated the evolution of DNA repair mechanisms that act specifically to counter the types of damage induced by oxidative stress.
Rate of evolutionary change
On some occasions, DNA damage is not repaired or is repaired by an error-prone mechanism that results in a change from the original sequence. When this occurs, mutations may propagate into the genomes of the cell's progeny. Should such an event occur in a germ line cell that will eventually produce a gamete, the mutation has the potential to be passed on to the organism's offspring. The rate of evolution in a particular species (or, in a particular gene) is a function of the rate of mutation. As a consequence, the rate and accuracy of DNA repair mechanisms have an influence over the process of evolutionary change.[130] DNA damage protection and repair does not influence the rate of adaptation by gene regulation and by recombination and selection of alleles. On the other hand, DNA damage repair and protection does influence the rate of accumulation of irreparable, advantageous, code expanding, inheritable mutations, and slows down the evolutionary mechanism for expansion of the genome of organisms with new functionalities. The tension between evolvability and mutation repair and protection needs further investigation.
Technology
A technology named clustered regularly interspaced short palindromic repeat (shortened to CRISPR-Cas9) was discovered in 2012. The new technology allows anyone with molecular biology training to alter the genes of any species with precision, by inducing DNA damage at a specific point and then altering DNA repair mechanisms to insert new genes.[131] It is cheaper, more efficient, and more precise than other technologies. With the help of CRISPR–Cas9, parts of a genome can be edited by scientists by removing, adding, or altering parts in a DNA sequence.
See also
- Accelerated aging disease
- Aging DNA
- Cell cycle
- DNA damage (naturally occurring)
- DNA damage theory of aging
- DNA replication
- Direct DNA damage
- Gene therapy
- Human mitochondrial genetics
- Indirect DNA damage
- Life extension
- Progeria
- REPAIRtoire
- Senescence
- SiDNA
- The scientific journal DNA Repair under Mutation Research
References
- "Nature Reviews Series: DNA damage". Nature Reviews Molecular Cell Biology. 5 July 2017. Retrieved 7 November 2018.
- Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J (2004). Molecular Biology of the Cell (5th ed.). New York: WH Freeman. p. 963.
- Acharya PV (1971). "The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides". Johns Hopkins Medical Journal. Supplement (1): 254–60. PMID 5055816.
- Bjorksten J, Acharya PV, Ashman S, Wetlaufer DB (July 1971). "Gerogenic fractions in the tritiated rat". Journal of the American Geriatrics Society. 19 (7): 561–74. doi:10.1111/j.1532-5415.1971.tb02577.x. PMID 5106728. S2CID 33154242.
- Browner WS, Kahn AJ, Ziv E, Reiner AP, Oshima J, Cawthon RM, et al. (December 2004). "The genetics of human longevity". The American Journal of Medicine. 117 (11): 851–60. CiteSeerX 10.1.1.556.6874. doi:10.1016/j.amjmed.2004.06.033. PMID 15589490.
- Broad WJ (7 October 2015). "Nobel Prize in Chemistry Awarded to Tomas Lindahl, Paul Modrich and Aziz Sancar for DNA Studies". The New York Times. Retrieved 7 October 2015.
- Staff (7 October 2015). "The Nobel Prize in Chemistry 2015 – DNA repair – providing chemical stability for life" (PDF). Nobel Prize. Retrieved 7 October 2015.
- Roulston A, Marcellus RC, Branton PE (1999). "Viruses and apoptosis". Annual Review of Microbiology. 53: 577–628. doi:10.1146/annurev.micro.53.1.577. PMID 10547702.
- Madigan MT, Martino JM (2006). Brock Biology of Microorganisms (11th ed.). Pearson. p. 136. ISBN 978-0-13-196893-6.
- Ohta T, Tokishita SI, Mochizuki K, Kawase J, Sakahira M, Yamagata H (2006). "UV Sensitivity and Mutagenesis of the Extremely Thermophilic Eubacterium Thermus thermophilus HB27". Genes and Environment. 28 (2): 56–61. doi:10.3123/jemsge.28.56.
- Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z (September 2006). "Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants". Cell Cycle. 5 (17): 1940–45. doi:10.4161/cc.5.17.3191. PMC 3488278. PMID 16940754.
- Braig M, Schmitt CA (March 2006). "Oncogene-induced senescence: putting the brakes on tumor development". Cancer Research. 66 (6): 2881–84. doi:10.1158/0008-5472.CAN-05-4006. PMID 16540631.
- Lynch MD (February 2006). "How does cellular senescence prevent cancer?". DNA and Cell Biology. 25 (2): 69–78. doi:10.1089/dna.2006.25.69. PMID 16460230.
- Campisi J, d'Adda di Fagagna F (September 2007). "Cellular senescence: when bad things happen to good cells". Nature Reviews. Molecular Cell Biology. 8 (9): 729–40. doi:10.1038/nrm2233. PMID 17667954. S2CID 15664931.
- Best BP (June 2009). "Nuclear DNA damage as a direct cause of aging" (PDF). Rejuvenation Research. 12 (3): 199–208. CiteSeerX 10.1.1.318.738. doi:10.1089/rej.2009.0847. PMID 19594328. Archived from the original (PDF) on 15 November 2017. Retrieved 29 September 2009.
- Sancar A (June 2003). "Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors". Chemical Reviews. 103 (6): 2203–37. doi:10.1021/cr0204348. PMID 12797829.
- Lucas-Lledó JI, Lynch M (May 2009). "Evolution of mutation rates: phylogenomic analysis of the photolyase/cryptochrome family". Molecular Biology and Evolution. 26 (5): 1143–53. doi:10.1093/molbev/msp029. PMC 2668831. PMID 19228922.
- Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2004). Molecular Biology of the Gene (5th ed.). Pearson Benjamin Cummings; CSHL Press. Ch. 9, 10. OCLC 936762772.
- Volkert MR (1988). "Adaptive response of Escherichia coli to alkylation damage". Environmental and Molecular Mutagenesis. 11 (2): 241–55. doi:10.1002/em.2850110210. PMID 3278898. S2CID 24722637.
- Willey J, Sherwood L, Woolverton C (2014). Prescott's Microbiology. New York: McGraw Hill. p. 381. ISBN 978-0-07-3402-40-6.
- Russell P (2018). i Genetics. Chennai: Pearson. p. 186. ISBN 978-93-325-7162-4.
- Reardon JT, Sancar A (2006). "Purification and characterization of Escherichia coli and human nucleotide excision repair enzyme systems". Methods in Enzymology. 408: 189–213. doi:10.1016/S0076-6879(06)08012-8. ISBN 9780121828134. PMID 16793370.
- Berg M, Tymoczko J, Stryer L (2012). Biochemistry 7th edition. New York: W.H. Freeman and Company. p. 840. ISBN 9781429229364.
- Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA (September 2005). "Modulation of DNA end joining by nuclear proteins". The Journal of Biological Chemistry. 280 (36): 31442–49. doi:10.1074/jbc.M503776200. PMID 16012167.
- Wilson TE, Grawunder U, Lieber MR (July 1997). "Yeast DNA ligase IV mediates non-homologous DNA end joining". Nature. 388 (6641): 495–98. Bibcode:1997Natur.388..495W. doi:10.1038/41365. PMID 9242411. S2CID 4422938.
- Moore JK, Haber JE (May 1996). "Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae". Molecular and Cellular Biology. 16 (5): 2164–73. doi:10.1128/mcb.16.5.2164. PMC 231204. PMID 8628283.
- Boulton SJ, Jackson SP (September 1996). "Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways". The EMBO Journal. 15 (18): 5093–103. doi:10.1002/j.1460-2075.1996.tb00890.x. PMC 452249. PMID 8890183.
- Wilson TE, Lieber MR (August 1999). "Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway". The Journal of Biological Chemistry. 274 (33): 23599–609. doi:10.1074/jbc.274.33.23599. PMID 10438542.
- Budman J, Chu G (February 2005). "Processing of DNA for nonhomologous end-joining by cell-free extract". The EMBO Journal. 24 (4): 849–60. doi:10.1038/sj.emboj.7600563. PMC 549622. PMID 15692565.
- Wang H, Perrault AR, Takeda Y, Qin W, Wang H, Iliakis G (September 2003). "Biochemical evidence for Ku-independent backup pathways of NHEJ". Nucleic Acids Research. 31 (18): 5377–88. doi:10.1093/nar/gkg728. PMC 203313. PMID 12954774.
- Jung D, Alt FW (January 2004). "Unraveling V(D)J recombination; insights into gene regulation". Cell. 116 (2): 299–311. doi:10.1016/S0092-8674(04)00039-X. PMID 14744439. S2CID 16890458.
- Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. (May 2013). "Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells". Proceedings of the National Academy of Sciences of the United States of America. 110 (19): 7720–25. Bibcode:2013PNAS..110.7720T. doi:10.1073/pnas.1213431110. PMC 3651503. PMID 23610439.
- Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC (March 2015). "Homology and enzymatic requirements of microhomology-dependent alternative end joining". Cell Death & Disease. 6 (3): e1697. doi:10.1038/cddis.2015.58. PMC 4385936. PMID 25789972.
- Decottignies A (2013). "Alternative end-joining mechanisms: a historical perspective". Frontiers in Genetics. 4: 48. doi:10.3389/fgene.2013.00048. PMC 3613618. PMID 23565119.
- Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, et al. (October 2006). "Reassembly of shattered chromosomes in Deinococcus radiodurans". Nature. 443 (7111): 569–73. Bibcode:2006Natur.443..569Z. doi:10.1038/nature05160. PMID 17006450. S2CID 4412830.
- Stenerlöw B, Karlsson KH, Cooper B, Rydberg B. "Measurement of prompt DNA double-strand breaks in mammalian cells without including heat-labile sites: results for cells deficient in nonhomologous end joining". Radiat Res. 2003 Apr;159(4):502–10. doi:10.1667/0033-7587(2003)159[0502:mopdds2.0.co;2] PMID 12643795.
- Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC (March 2009). "Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance". Microbiology and Molecular Biology Reviews. 73 (1): 134–54. doi:10.1128/MMBR.00034-08. PMC 2650891. PMID 19258535.
- Colis LC, Raychaudhury P, Basu AK (August 2008). "Mutational specificity of gamma-radiation-induced guanine-thymine and thymine-guanine intrastrand cross-links in mammalian cells and translesion synthesis past the guanine-thymine lesion by human DNA polymerase eta". Biochemistry. 47 (31): 8070–79. doi:10.1021/bi800529f. PMC 2646719. PMID 18616294.
- Raychaudhury P, Basu AK (March 2011). "Genetic requirement for mutagenesis of the G[8,5-Me]T cross-link in Escherichia coli: DNA polymerases IV and V compete for error-prone bypass". Biochemistry. 50 (12): 2330–38. doi:10.1021/bi102064z. PMC 3062377. PMID 21302943.
- "Translesion Synthesis". Research.chem.psu.edu. Archived from the original on 10 March 2012. Retrieved 14 August 2012.
- Wang Z (July 2001). "Translesion synthesis by the UmuC family of DNA polymerases". Mutation Research. 486 (2): 59–70. doi:10.1016/S0921-8777(01)00089-1. PMID 11425512.
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. (2006). DNA Repair and Mutagenesis, part 3. ASM Press. 2nd ed.
- Liu B, Yip RK, Zhou Z (November 2012). "Chromatin remodeling, DNA damage repair and aging". Current Genomics. 13 (7): 533–47. doi:10.2174/138920212803251373. PMC 3468886. PMID 23633913.
- Halicka HD, Zhao H, Podhorecka M, Traganos F, Darzynkiewicz Z (July 2009). "Cytometric detection of chromatin relaxation, an early reporter of DNA damage response". Cell Cycle. 8 (14): 2233–37. doi:10.4161/cc.8.14.8984. PMC 3856216. PMID 19502789.
- Sellou H, Lebeaupin T, Chapuis C, Smith R, Hegele A, Singh HR, et al. (December 2016). "The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage". Molecular Biology of the Cell. 27 (24): 3791–99. doi:10.1091/mbc.E16-05-0269. PMC 5170603. PMID 27733626.
- Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, et al. (September 2016). "JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks". Cell Reports. 16 (10): 2641–50. doi:10.1016/j.celrep.2016.08.006. PMC 5089070. PMID 27568560.
- Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (January 2008). "PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites". The Journal of Biological Chemistry. 283 (2): 1197–208. doi:10.1074/jbc.M706734200. PMID 18025084.
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (March 1998). "DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139". The Journal of Biological Chemistry. 273 (10): 5858–68. doi:10.1074/jbc.273.10.5858. PMID 9488723.
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (November 2007). "RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins". Cell. 131 (5): 887–900. doi:10.1016/j.cell.2007.09.040. PMID 18001824. S2CID 14232192.
- Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH, et al. (May 2012). "A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure". The EMBO Journal. 31 (11): 2511–27. doi:10.1038/emboj.2012.104. PMC 3365417. PMID 22531782.
- Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, et al. (August 2007). "Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC". Journal of Cell Science. 120 (Pt 15): 2706–16. doi:10.1242/jcs.008367. PMID 17635991.
- Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, et al. (October 2012). "PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1". The Journal of Cell Biology. 199 (2): 235–49. doi:10.1083/jcb.201112132. PMC 3471223. PMID 23045548.
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP (January 2006). "ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks". Nature Cell Biology. 8 (1): 37–45. doi:10.1038/ncb1337. PMID 16327781. S2CID 9797133.
- Bakkenist CJ, Kastan MB (January 2003). "DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation". Nature. 421 (6922): 499–506. Bibcode:2003Natur.421..499B. doi:10.1038/nature01368. PMID 12556884. S2CID 4403303.
- Wei Q, Li L, Chen D (2007). DNA Repair, Genetic Instability, and Cancer. World Scientific. ISBN 978-981-270-014-8.
- Schonthal AH (2004). Checkpoint Controls and Cancer. Humana Press. ISBN 978-1-58829-500-2.
- Gartel AL, Tyner AL (June 2002). "The role of the cyclin-dependent kinase inhibitor p21 in apoptosis". Molecular Cancer Therapeutics. 1 (8): 639–49. PMID 12479224.
- Janion C (2001). "Some aspects of the SOS response system--a critical survey". Acta Biochimica Polonica. 48 (3): 599–610. doi:10.18388/abp.2001_3894. PMID 11833768.
- Erill I, Campoy S, Barbé J (November 2007). "Aeons of distress: an evolutionary perspective on the bacterial SOS response". FEMS Microbiology Reviews. 31 (6): 637–56. doi:10.1111/j.1574-6976.2007.00082.x. PMID 17883408.
- Schlacher K, Pham P, Cox MM, Goodman MF (February 2006). "Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation". Chemical Reviews. 106 (2): 406–19. doi:10.1021/cr0404951. PMID 16464012.
- Fry RC, Begley TJ, Samson LD (2004). "Genome-wide responses to DNA-damaging agents". Annual Review of Microbiology. 59: 357–77. doi:10.1146/annurev.micro.59.031805.133658. PMID 16153173.
- Espejel S, Martín M, Klatt P, Martín-Caballero J, Flores JM, Blasco MA (May 2004). "Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice". EMBO Reports. 5 (5): 503–09. doi:10.1038/sj.embor.7400127. PMC 1299048. PMID 15105825.
- de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, et al. (May 2002). "Premature aging in mice deficient in DNA repair and transcription". Science. 296 (5571): 1276–79. Bibcode:2002Sci...296.1276D. doi:10.1126/science.1070174. PMID 11950998. S2CID 41930529.
- Dollé ME, Busuttil RA, Garcia AM, Wijnhoven S, van Drunen E, Niedernhofer LJ, et al. (April 2006). "Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice". Mutation Research. 596 (1–2): 22–35. doi:10.1016/j.mrfmmm.2005.11.008. PMID 16472827.
- Kobayashi Y, Narumi I, Satoh K, Funayama T, Kikuchi M, Kitayama S, Watanabe H (November 2004). "Radiation response mechanisms of the extremely radioresistant bacterium Deinococcus radiodurans". Uchu Seibutsu Kagaku. 18 (3): 134–35. PMID 15858357.
- Spindler SR (September 2005). "Rapid and reversible induction of the longevity, anticancer and genomic effects of caloric restriction". Mechanisms of Ageing and Development. 126 (9): 960–66. doi:10.1016/j.mad.2005.03.016. PMID 15927235. S2CID 7067036.
- Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z (December 2012). "Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling". Aging. 4 (12): 952–65. doi:10.18632/aging.100521. PMC 3615161. PMID 23363784.
- Tissenbaum HA, Guarente L (March 2001). "Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans". Nature. 410 (6825): 227–30. Bibcode:2001Natur.410..227T. doi:10.1038/35065638. PMID 11242085. S2CID 4356885.
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. (July 2004). "Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase". Science. 305 (5682): 390–92. Bibcode:2004Sci...305..390C. doi:10.1126/science.1099196. PMID 15205477. S2CID 33503081.
- Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A, Heydari AR (March 2003). "Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline". DNA Repair. 2 (3): 295–307. doi:10.1016/S1568-7864(02)00219-7. PMID 12547392.
- Stuart JA, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA (March 2004). "Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction". FASEB Journal. 18 (3): 595–97. doi:10.1096/fj.03-0890fje. PMID 14734635. S2CID 43118901.
- Walker DW, McColl G, Jenkins NL, Harris J, Lithgow GJ (May 2000). "Evolution of lifespan in C. elegans". Nature. 405 (6784): 296–97. doi:10.1038/35012693. PMID 10830948. S2CID 4402039.
- Johnson G (28 December 2010). "Unearthing Prehistoric Tumors, and Debate". The New York Times.
If we lived long enough, sooner or later we all would get cancer.
- Alberts B, Johnson A, Lewis J, et al. (2002). "The Preventable Causes of Cancer". Molecular biology of the cell (4th ed.). New York: Garland Science. ISBN 978-0-8153-4072-0.
A certain irreducible background incidence of cancer is to be expected regardless of circumstances: mutations can never be absolutely avoided, because they are an inescapable consequence of fundamental limitations on the accuracy of DNA replication, as discussed in Chapter 5. If a human could live long enough, it is inevitable that at least one of his or her cells would eventually accumulate a set of mutations sufficient for cancer to develop.
- Friedenson B (August 2007). "The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers". BMC Cancer. 7: 152. doi:10.1186/1471-2407-7-152. PMC 1959234. PMID 17683622.
- Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, Turchi JJ (April 2016). "DNA repair targeted therapy: The past or future of cancer treatment?". Pharmacology & Therapeutics. 160: 65–83. doi:10.1016/j.pharmthera.2016.02.003. PMC 4811676. PMID 26896565.
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. (April 2005). "Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase". Nature. 434 (7035): 913–17. Bibcode:2005Natur.434..913B. doi:10.1038/nature03443. PMID 15829966. S2CID 4391043.
- Goldstein M, Kastan MB (2015). "The DNA damage response: implications for tumor responses to radiation and chemotherapy". Annual Review of Medicine. 66: 129–43. doi:10.1146/annurev-med-081313-121208. PMID 25423595.
- Jeggo PA, Pearl LH, Carr AM (January 2016). "DNA repair, genome stability and cancer: a historical perspective" (PDF). Nature Reviews. Cancer. 16 (1): 35–42. doi:10.1038/nrc.2015.4. PMID 26667849. S2CID 14941857.
- Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, et al. (April 2005). "DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis". Nature. 434 (7035): 864–70. Bibcode:2005Natur.434..864B. doi:10.1038/nature03482. PMID 15829956. S2CID 4398393.
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. (November 2006). "Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints". Nature. 444 (7119): 633–37. Bibcode:2006Natur.444..633B. doi:10.1038/nature05268. PMID 17136093. S2CID 4406956.
- Gaillard H, García-Muse T, Aguilera A (May 2015). "Replication stress and cancer". Nature Reviews. Cancer. 15 (5): 276–89. doi:10.1038/nrc3916. hdl:10261/123721. PMID 25907220. S2CID 11342123.
- Halazonetis TD, Gorgoulis VG, Bartek J (March 2008). "An oncogene-induced DNA damage model for cancer development". Science. 319 (5868): 1352–55. Bibcode:2008Sci...319.1352H. doi:10.1126/science.1140735. PMID 18323444. S2CID 16426080.
- de Boer J, Hoeijmakers JH (March 2000). "Nucleotide excision repair and human syndromes" (PDF). Carcinogenesis. 21 (3): 453–60. doi:10.1093/carcin/21.3.453. PMID 10688865.
- Broustas CG, Lieberman HB (February 2014). "DNA damage response genes and the development of cancer metastasis". Radiation Research. 181 (2): 111–30. Bibcode:2014RadR..181..111B. doi:10.1667/RR13515.1. PMC 4064942. PMID 24397478.
- Zhang P, Wang J, Gao W, Yuan BZ, Rogers J, Reed E (May 2004). "CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer". Molecular Cancer. 3 (4): 14. doi:10.1186/1476-4598-3-14. PMC 419366. PMID 15125777.
- Baylin SB, Ohm JE (February 2006). "Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?". Nature Reviews. Cancer. 6 (2): 107–16. doi:10.1038/nrc1799. PMID 16491070. S2CID 2514545.
- Kanwal R, Gupta S (April 2012). "Epigenetic modifications in cancer". Clinical Genetics. 81 (4): 303–11. doi:10.1111/j.1399-0004.2011.01809.x. PMC 3590802. PMID 22082348.
- Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, et al. (April 2003). "Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma". Molecular and Cellular Biology. 23 (7): 2225–38. doi:10.1128/MCB.23.7.2225-2238.2003. PMC 150734. PMID 12640109.
- Jacinto FV, Esteller M (July 2007). "Mutator pathways unleashed by epigenetic silencing in human cancer". Mutagenesis. 22 (4): 247–53. doi:10.1093/mutage/gem009. PMID 17412712.
- Lahtz C, Pfeifer GP (February 2011). "Epigenetic changes of DNA repair genes in cancer". Journal of Molecular Cell Biology. 3 (1): 51–58. doi:10.1093/jmcb/mjq053. PMC 3030973. PMID 21278452. Epigenetic changes of DNA repair genes in cancer
- Bernstein C, Nfonsam V, Prasad AR, Bernstein H (March 2013). "Epigenetic field defects in progression to cancer". World Journal of Gastrointestinal Oncology. 5 (3): 43–49. doi:10.4251/wjgo.v5.i3.43. PMC 3648662. PMID 23671730.
- Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proceedings of the National Academy of Sciences of the United States of America. 94 (7): 3122–27. Bibcode:1997PNAS...94.3122N. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (December 2006). "Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6". Carcinogenesis. 27 (12): 2402–08. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (March 2002). "Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation". EMBO Reports. 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- German J (March 1969). "Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients". American Journal of Human Genetics. 21 (2): 196–227. PMC 1706430. PMID 5770175.
- O'Hagan HM, Mohammad HP, Baylin SB (August 2008). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLOS Genetics. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, et al. (July 2007). "DNA damage, homology-directed repair, and DNA methylation". PLOS Genetics. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- Malkin D (April 2011). "Li-fraumeni syndrome". Genes & Cancer. 2 (4): 475–84. doi:10.1177/1947601911413466. PMC 3135649. PMID 21779515.
- Fearon ER (November 1997). "Human cancer syndromes: clues to the origin and nature of cancer". Science. 278 (5340): 1043–50. Bibcode:1997Sci...278.1043F. doi:10.1126/science.278.5340.1043. PMID 9353177.
- Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (June 2005). "O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions". Gut. 54 (6): 797–802. doi:10.1136/gut.2004.059535. PMC 1774551. PMID 15888787.
- Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, et al. (September 2005). "MGMT promoter methylation and field defect in sporadic colorectal cancer". Journal of the National Cancer Institute. 97 (18): 1330–38. doi:10.1093/jnci/dji275. PMID 16174854.
- Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K, Kolios G (July 2010). "Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas". World Journal of Gastroenterology. 16 (28): 3553–60. doi:10.3748/wjg.v16.i28.3553. PMC 2909555. PMID 20653064.
- Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, et al. (October 2011). "Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence". Langenbeck's Archives of Surgery. 396 (7): 1017–26. doi:10.1007/s00423-011-0812-9. PMID 21706233. S2CID 8069716.
- Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, et al. (April 2013). "Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer". Clinical Cancer Research. 19 (8): 2265–72. doi:10.1158/1078-0432.CCR-12-3518. PMID 23422094.
- Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, Hosseini SV (May 2013). "Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer". Molecular Biology Reports. 40 (5): 3851–57. doi:10.1007/s11033-012-2465-3. PMID 23271133. S2CID 18733871.
- Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. (May 2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology. 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, et al. (April 2010). "Modulation of mismatch repair and genomic stability by miR-155". Proceedings of the National Academy of Sciences of the United States of America. 107 (15): 6982–87. Bibcode:2010PNAS..107.6982V. doi:10.1073/pnas.1002472107. PMC 2872463. PMID 20351277.
- Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, et al. (April 2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integrity. 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- Human DNA Repair Genes, 15 April 2014, MD Anderson Cancer Center, University of Texas
- Jin B, Robertson KD (2013). "DNA methyltransferases, DNA damage repair, and cancer". Advances in Experimental Medicine and Biology. 754: 3–29. doi:10.1007/978-1-4419-9967-2_1. ISBN 978-1-4419-9966-5. PMC 3707278. PMID 22956494.
- Krishnan K, Steptoe AL, Martin HC, Wani S, Nones K, Waddell N, et al. (February 2013). "MicroRNA-182-5p targets a network of genes involved in DNA repair". RNA. 19 (2): 230–42. doi:10.1261/rna.034926.112. PMC 3543090. PMID 23249749.
- Chaisaingmongkol J, Popanda O, Warta R, Dyckhoff G, Herpel E, Geiselhart L, et al. (December 2012). "Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma". Oncogene. 31 (49): 5108–16. doi:10.1038/onc.2011.660. PMID 22286769.
- Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, et al. (November 2008). "Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers". Molecular Cancer Research. 6 (11): 1710–17. doi:10.1158/1541-7786.MCR-08-0269. PMC 2948671. PMID 19010819.
- Lam JS, Seligson DB, Yu H, Li A, Eeva M, Pantuck AJ, et al. (August 2006). "Flap endonuclease 1 is overexpressed in prostate cancer and is associated with a high Gleason score". BJU International. 98 (2): 445–51. doi:10.1111/j.1464-410X.2006.06224.x. PMID 16879693. S2CID 22165252.
- Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, et al. (January 2005). "Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells". Clinical Cancer Research. 11 (2 Pt 1): 473–82. doi:10.1158/1078-0432.473.11.2. PMID 15701830.
- Wang K, Xie C, Chen D (May 2014). "Flap endonuclease 1 is a promising candidate biomarker in gastric cancer and is involved in cell proliferation and apoptosis". International Journal of Molecular Medicine. 33 (5): 1268–74. doi:10.3892/ijmm.2014.1682. PMID 24590400.
- Krause A, Combaret V, Iacono I, Lacroix B, Compagnon C, Bergeron C, et al. (July 2005). "Genome-wide analysis of gene expression in neuroblastomas detected by mass screening" (PDF). Cancer Letters. 225 (1): 111–20. doi:10.1016/j.canlet.2004.10.035. PMID 15922863. S2CID 44644467.
- Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, van Heek NT, Rosty C, et al. (April 2003). "Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays". The American Journal of Pathology. 162 (4): 1151–62. doi:10.1016/S0002-9440(10)63911-9. PMC 1851213. PMID 12651607.
- Sato M, Girard L, Sekine I, Sunaga N, Ramirez RD, Kamibayashi C, Minna JD (October 2003). "Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer". Oncogene. 22 (46): 7243–46. doi:10.1038/sj.onc.1206977. PMID 14562054.
- Bi FF, Li D, Yang Q (2013). "Hypomethylation of ETS transcription factor binding sites and upregulation of PARP1 expression in endometrial cancer". BioMed Research International. 2013: 946268. doi:10.1155/2013/946268. PMC 3666359. PMID 23762867.
- Bi FF, Li D, Yang Q (February 2013). "Promoter hypomethylation, especially around the E26 transformation-specific motif, and increased expression of poly (ADP-ribose) polymerase 1 in BRCA-mutated serous ovarian cancer". BMC Cancer. 13: 90. doi:10.1186/1471-2407-13-90. PMC 3599366. PMID 23442605.
- Supek F, Lehner B (May 2015). "Differential DNA mismatch repair underlies mutation rate variation across the human genome". Nature. 521 (7550): 81–84. Bibcode:2015Natur.521...81S. doi:10.1038/nature14173. PMC 4425546. PMID 25707793.
- Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, et al. (November 2014). "Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes". Cell Reports. 9 (4): 1228–34. doi:10.1016/j.celrep.2014.10.031. PMC 4254608. PMID 25456125.
- Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM (April 2013). "The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα". Cell. 153 (3): 590–600. doi:10.1016/j.cell.2013.03.025. PMC 3641580. PMID 23622243.
- Supek F, Lehner B (July 2017). "Clustered Mutation Signatures Reveal that Error-Prone DNA Repair Targets Mutations to Active Genes". Cell. 170 (3): 534–547.e23. doi:10.1016/j.cell.2017.07.003. hdl:10230/35343. PMID 28753428.
- Polak P, Lawrence MS, Haugen E, Stoletzki N, Stojanov P, Thurman RE, et al. (January 2014). "Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair". Nature Biotechnology. 32 (1): 71–75. doi:10.1038/nbt.2778. PMC 4116484. PMID 24336318.
- Cromie GA, Connelly JC, Leach DR (December 2001). "Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans". Molecular Cell. 8 (6): 1163–74. doi:10.1016/S1097-2765(01)00419-1. PMID 11779493.
- O'Brien PJ (February 2006). "Catalytic promiscuity and the divergent evolution of DNA repair enzymes". Chemical Reviews. 106 (2): 720–52. doi:10.1021/cr040481v. PMID 16464022.
- Maresca B, Schwartz JH (January 2006). "Sudden origins: a general mechanism of evolution based on stress protein concentration and rapid environmental change". The Anatomical Record Part B: The New Anatomist. 289 (1): 38–46. doi:10.1002/ar.b.20089. PMID 16437551.
- "CRISPR gene-editing tool has scientists thrilled – but nervous" CBC news. Author Kelly Crowe. 30 November 2015.
External links
| Library resources about DNA repair |
 Media related to DNA repair at Wikimedia Commons
Media related to DNA repair at Wikimedia Commons- Roswell Park Cancer Institute DNA Repair Lectures
- A comprehensive list of Human DNA Repair Genes
- 3D structures of some DNA repair enzymes
- Human DNA repair diseases
- DNA repair special interest group
- DNA Repair Archived 12 February 2018 at the Wayback Machine
- DNA Damage and DNA Repair
- Segmental Progeria
- DNA-damage repair; the good, the bad, and the ugly