Autoimmunity
Autoimmunity is the system of immune responses of an organism against its own healthy cells, tissues and other body normal constituents.[1][2] Any disease resulting from this type of immune response is termed an "autoimmune disease". Prominent examples include celiac disease, post-infectious IBS, diabetes mellitus type 1, Henloch Scholein Pupura (HSP) sarcoidosis, systemic lupus erythematosus (SLE), Sjögren syndrome, eosinophilic granulomatosis with polyangiitis, Hashimoto's thyroiditis, Graves' disease, idiopathic thrombocytopenic purpura, Addison's disease, rheumatoid arthritis (RA), ankylosing spondylitis, polymyositis (PM), dermatomyositis (DM), Alopecia Areata [3] and multiple sclerosis (MS). Autoimmune diseases are very often treated with steroids.[4]
| Autoimmunity | |
|---|---|
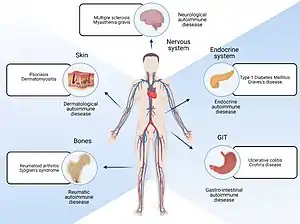 | |
| Parts of body affected by autoimmune diseases | |
| Specialty | Immunology |
Autoimmunity means presence of antibodies or T cells that react with self-protein and is present in all individuals, even in normal health state. It causes autoimmune diseases if self-reactivity can lead to tissue damage.[5]
History
In the later 19th century it was believed that the immune system was unable to react against the body's own tissues. Paul Ehrlich, at the turn of the 20th century, proposed the concept of horror autotoxicus. Ehrlich later adjusted his theory to recognize the possibility of autoimmune tissue attacks, but believed certain innate protection mechanisms would prevent the autoimmune response from becoming pathological.
In 1904 this theory was challenged by the discovery of a substance in the serum of patients with paroxysmal cold hemoglobinuria that reacted with red blood cells. During the following decades, a number of conditions could be linked to autoimmune responses. However, the authoritative status of Ehrlich's postulate hampered the understanding of these findings. Immunology became a biochemical rather than a clinical discipline.[6] By the 1950s the modern understanding of autoantibodies and autoimmune diseases started to spread.
More recently it has become accepted that autoimmune responses are an integral part of vertebrate immune systems (sometimes termed "natural autoimmunity").[7] Autoimmunity should not be confused with alloimmunity.
Low-level autoimmunity
While a high level of autoimmunity is unhealthy, a low level of autoimmunity may actually be beneficial. Taking the experience of a beneficial factor in autoimmunity further, one might hypothesize with intent to prove that autoimmunity is always a self-defense mechanism of the mammal system to survive. The system does not randomly lose the ability to distinguish between self and non-self; the attack on cells may be the consequence of cycling metabolic processes necessary to keep the blood chemistry in homeostasis.
Second, autoimmunity may have a role in allowing a rapid immune response in the early stages of an infection when the availability of foreign antigens limits the response (i.e., when there are few pathogens present). In their study, Stefanova et al. (2002) injected an anti-MHC class II antibody into mice expressing a single type of MHC Class II molecule (H-2b) to temporarily prevent CD4+ T cell-MHC interaction. Naive CD4+ T cells (those that have not encountered non-self antigens before) recovered from these mice 36 hours post-anti-MHC administration showed decreased responsiveness to the antigen pigeon cytochrome c peptide, as determined by ZAP70 phosphorylation, proliferation, and interleukin 2 production. Thus Stefanova et al. (2002) demonstrated that self-MHC recognition (which, if too strong may contribute to autoimmune disease) maintains the responsiveness of CD4+ T cells when foreign antigens are absent.[8]
Immunological tolerance
Pioneering work by Noel Rose and Ernst Witebsky in New York, and Roitt and Doniach at University College London provided clear evidence that, at least in terms of antibody-producing B cells (B lymphocytes), diseases such as rheumatoid arthritis and thyrotoxicosis are associated with loss of immunological tolerance, which is the ability of an individual to ignore "self", while reacting to "non-self". This breakage leads to the immune system mounting an effective and specific immune response against self antigens. The exact genesis of immunological tolerance is still elusive, but several theories have been proposed since the mid-twentieth century to explain its origin.
Three hypotheses have gained widespread attention among immunologists:
- Clonal deletion theory, proposed by Burnet, according to which self-reactive lymphoid cells are destroyed during the development of the immune system in an individual. For their work Frank M. Burnet and Peter B. Medawar were awarded the 1960 Nobel Prize in Physiology or Medicine "for discovery of acquired immunological tolerance".
- Clonal anergy theory, proposed by Nossal, in which self-reactive T- or B-cells become inactivated in the normal individual and cannot amplify the immune response.[9]
- Idiotype network theory, proposed by Jerne, wherein a network of antibodies capable of neutralizing self-reactive antibodies exists naturally within the body.[10]
In addition, two other theories are under intense investigation:
- Clonal ignorance theory, according to which autoreactive T cells that are not represented in the thymus will mature and migrate to the periphery, where they will not encounter the appropriate antigen because it is inaccessible tissues. Consequently, auto-reactive B cells, that escape deletion, cannot find the antigen or the specific helper T cell.[11]
- Suppressor population or Regulatory T cell theory, wherein regulatory T-lymphocytes (commonly CD4+FoxP3+ cells, among others) function to prevent, downregulate, or limit autoaggressive immune responses in the immune system.
Tolerance can also be differentiated into "central" and "peripheral" tolerance, on whether or not the above-stated checking mechanisms operate in the central lymphoid organs (thymus and bone marrow) or the peripheral lymphoid organs (lymph node, spleen, etc., where self-reactive B-cells may be destroyed). It must be emphasised that these theories are not mutually exclusive, and evidence has been mounting suggesting that all of these mechanisms may actively contribute to vertebrate immunological tolerance.
A puzzling feature of the documented loss of tolerance seen in spontaneous human autoimmunity is that it is almost entirely restricted to the autoantibody responses produced by B lymphocytes. Loss of tolerance by T cells has been extremely hard to demonstrate, and where there is evidence for an abnormal T cell response it is usually not to the antigen recognised by autoantibodies. Thus, in rheumatoid arthritis there are autoantibodies to IgG Fc but apparently no corresponding T cell response. In systemic lupus there are autoantibodies to DNA, which cannot evoke a T cell response, and limited evidence for T cell responses implicates nucleoprotein antigens. In Celiac disease there are autoantibodies to tissue transglutaminase but the T cell response is to the foreign protein gliadin. This disparity has led to the idea that human autoimmune disease is in most cases (with probable exceptions including type I diabetes) based on a loss of B cell tolerance which makes use of normal T cell responses to foreign antigens in a variety of aberrant ways.[12]
Immunodeficiency and autoimmunity
There are a large number of immunodeficiency syndromes that present clinical and laboratory characteristics of autoimmunity. The decreased ability of the immune system to clear infections in these patients may be responsible for causing autoimmunity through perpetual immune system activation.[13]
One example is common variable immunodeficiency (CVID) where multiple autoimmune diseases are seen, e.g.: inflammatory bowel disease, autoimmune thrombocytopenia and autoimmune thyroid disease.
Familial hemophagocytic lymphohistiocytosis, an autosomal recessive primary immunodeficiency, is another example. Pancytopenia, rashes, swollen lymph nodes and enlargement of the liver and spleen are commonly seen in such individuals. Presence of multiple uncleared viral infections due to lack of perforin are thought to be responsible.
In addition to chronic and/or recurrent infections many autoimmune diseases including arthritis, autoimmune hemolytic anemia, scleroderma and type 1 diabetes mellitus are also seen in X-linked agammaglobulinemia (XLA). Recurrent bacterial and fungal infections and chronic inflammation of the gut and lungs are seen in chronic granulomatous disease (CGD) as well. CGD is a caused by decreased production of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase by neutrophils. Hypomorphic RAG mutations are seen in patients with midline granulomatous disease; an autoimmune disorder that is commonly seen in patients with granulomatosis with polyangiitis and NK/T cell lymphomas.Wiskott–Aldrich syndrome (WAS) patients also present with eczema, autoimmune manifestations, recurrent bacterial infections and lymphoma. In autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy also autoimmunity and infections coexist: organ-specific autoimmune manifestations (e.g., hypoparathyroidism and adrenocortical failure) and chronic mucocutaneous candidiasis. Finally, IgA deficiency is also sometimes associated with the development of autoimmune and atopic phenomena.
Genetic factors
Certain individuals are genetically susceptible to developing autoimmune diseases. This susceptibility is associated with multiple genes plus other risk factors. Genetically predisposed individuals do not always develop autoimmune diseases. Three main sets of genes are suspected in many autoimmune diseases. These genes are related to:
- Immunoglobulins
- T-cell receptors
- The major histocompatibility complexes (MHC).
The first two, which are involved in the recognition of antigens, are inherently variable and susceptible to recombination. These variations enable the immune system to respond to a very wide variety of invaders, but may also give rise to lymphocytes capable of self-reactivity.
- HLA DR2 is strongly positively correlated with systemic lupus erythematosus, narcolepsy[14] and multiple sclerosis, and negatively correlated with DM Type 1.
- HLA DR3 is correlated strongly with Sjögren syndrome, myasthenia gravis, SLE, and DM Type 1.
- HLA DR4 is correlated with the genesis of rheumatoid arthritis, Type 1 diabetes mellitus, and pemphigus vulgaris.
Fewer correlations exist with MHC class I molecules. The most notable and consistent is the association between HLA B27 and spondyloarthropathies like ankylosing spondylitis and reactive arthritis. Correlations may exist between polymorphisms within class II MHC promoters and autoimmune disease.
The contributions of genes outside the MHC complex remain the subject of research, in animal models of disease (Linda Wicker's extensive genetic studies of diabetes in the NOD mouse), and in patients (Brian Kotzin's linkage analysis of susceptibility to SLE).
Recently, PTPN22 has been associated with multiple autoimmune diseases including Type I diabetes, rheumatoid arthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, Graves' disease, Addison's disease, Myasthenia Gravis, vitiligo, systemic sclerosis juvenile idiopathic arthritis, and psoriatic arthritis.[15]
Sex
| Ratio of female/male incidence of autoimmune diseases | |
|---|---|
| Hashimoto's thyroiditis | 10:1[16] |
| Graves' disease | 7:1[16] |
| Multiple sclerosis (MS) | 2:1[16] |
| Myasthenia gravis | 2:1[16] |
| Systemic lupus erythematosus (SLE) | 9:1[16] |
| Rheumatoid arthritis | 5:2[16] |
| Primary sclerosing cholangitis | 1:2 |
There is some evidence that a person's sex may also have some role in the development of autoimmunity; that is, most autoimmune diseases are sex-related. A few autoimmune diseases that men are just as or more likely to develop as women include: ankylosing spondylitis, type 1 diabetes mellitus, granulomatosis with polyangiitis, Crohn's disease, Primary sclerosing cholangitis and psoriasis.
The reasons for the sex role in autoimmunity vary. Women appear to generally mount larger inflammatory responses than men when their immune systems are triggered, increasing the risk of autoimmunity. Involvement of sex steroids is indicated by that many autoimmune diseases tend to fluctuate in accordance with hormonal changes, for example: during pregnancy, in the menstrual cycle, or when using oral contraception. A history of pregnancy also appears to leave a persistent increased risk for autoimmune disease. It has been suggested that the slight, direct exchange of cells between mothers and their children during pregnancy may induce autoimmunity.[17] This would tip the gender balance in the direction of the female.
Another theory suggests the female high tendency to get autoimmunity is due to an imbalanced X-chromosome inactivation.[18] The X-inactivation skew theory, proposed by Princeton University's Jeff Stewart, has recently been confirmed experimentally in scleroderma and autoimmune thyroiditis.[19] Other complex X-linked genetic susceptibility mechanisms are proposed and under investigation.
Environmental factors
Infectious diseases and parasites
An interesting inverse relationship exists between infectious diseases and autoimmune diseases. In areas where multiple infectious diseases are endemic, autoimmune diseases are quite rarely seen. The reverse, to some extent, seems to hold true. The hygiene hypothesis attributes these correlations to the immune-manipulating strategies of pathogens. While such an observation has been variously termed as spurious and ineffective, according to some studies, parasite infection is associated with reduced activity of autoimmune disease.[20][21][22]
The putative mechanism is that the parasite attenuates the host immune response in order to protect itself. This may provide a serendipitous benefit to a host that also has autoimmune disease. The details of parasite immune modulation are not yet known, but may include secretion of anti-inflammatory agents or interference with the host immune signaling.
A paradoxical observation has been the strong association of certain microbial organisms with autoimmune diseases. For example, Klebsiella pneumoniae and coxsackievirus B have been strongly correlated with ankylosing spondylitis and diabetes mellitus type 1, respectively. This has been explained by the tendency of the infecting organism to produce super-antigens that are capable of polyclonal activation of B-lymphocytes, and production of large amounts of antibodies of varying specificities, some of which may be self-reactive (see below).
Chemical agents and drugs
Certain chemical agents and drugs can also be associated with the genesis of autoimmune conditions, or conditions that simulate autoimmune diseases. The most striking of these is the drug-induced lupus erythematosus. Usually, withdrawal of the offending drug cures the symptoms in a patient.
Cigarette smoking is now established as a major risk factor for both incidence and severity of rheumatoid arthritis. This may relate to abnormal citrullination of proteins, since the effects of smoking correlate with the presence of antibodies to citrullinated peptides.
Pathogenesis of autoimmunity
Several mechanisms are thought to be operative in the pathogenesis of autoimmune diseases, against a backdrop of genetic predisposition and environmental modulation. It is beyond the scope of this article to discuss each of these mechanisms exhaustively, but a summary of some of the important mechanisms have been described:
- T-cell bypass – A normal immune system requires the activation of B cells by T cells before the former can undergo differentiation into plasma B-cells and subsequently produce antibodies in large quantities. This requirement of a T cell can be bypassed in rare instances, such as infection by organisms producing super-antigens, which are capable of initiating polyclonal activation of B-cells, or even of T-cells, by directly binding to the β-subunit of T-cell receptors in a non-specific fashion.
- T-cell–B-cell discordance – A normal immune response is assumed to involve B and T cell responses to the same antigen, even if we know that B cells and T cells recognise very different things: conformations on the surface of a molecule for B cells and pre-processed peptide fragments of proteins for T cells. However, there is nothing as far as we know that requires this. All that is required is that a B cell recognising antigen X endocytoses and processes a protein Y (normally =X) and presents it to a T cell. Roosnek and Lanzavecchia showed that B cells recognising IgGFc could get help from any T cell responding to an antigen co-endocytosed with IgG by the B cell as part of an immune complex. In coeliac disease it seems likely that B cells recognising tissue transglutamine are helped by T cells recognising gliadin.
- Aberrant B cell receptor-mediated feedback – A feature of human autoimmune disease is that it is largely restricted to a small group of antigens, several of which have known signaling roles in the immune response (DNA, C1q, IgGFc, Ro, Con. A receptor, Peanut agglutinin receptor(PNAR)). This fact gave rise to the idea that spontaneous autoimmunity may result when the binding of antibody to certain antigens leads to aberrant signals being fed back to parent B cells through membrane bound ligands. These ligands include B cell receptor (for antigen), IgG Fc receptors, CD21, which binds complement C3d, Toll-like receptors 9 and 7 (which can bind DNA and nucleoproteins) and PNAR. More indirect aberrant activation of B cells can also be envisaged with autoantibodies to acetyl choline receptor (on thymic myoid cells) and hormone and hormone binding proteins. Together with the concept of T-cell–B-cell discordance this idea forms the basis of the hypothesis of self-perpetuating autoreactive B cells.[23] Autoreactive B cells in spontaneous autoimmunity are seen as surviving because of subversion both of the T cell help pathway and of the feedback signal through B cell receptor, thereby overcoming the negative signals responsible for B cell self-tolerance without necessarily requiring loss of T cell self-tolerance.
- Molecular mimicry – An exogenous antigen may share structural similarities with certain host antigens; thus, any antibody produced against this antigen (which mimics the self-antigens) can also, in theory, bind to the host antigens, and amplify the immune response. The idea of molecular mimicry arose in the context of rheumatic fever, which follows infection with Group A beta-haemolytic streptococci. Although rheumatic fever has been attributed to molecular mimicry for half a century no antigen has been formally identified (if anything too many have been proposed). Moreover, the complex tissue distribution of the disease (heart, joint, skin, basal ganglia) argues against a cardiac specific antigen. It remains entirely possible that the disease is due to e.g. an unusual interaction between immune complexes, complement components and endothelium.
- Idiotype cross-reaction – Idiotypes are antigenic epitopes found in the antigen-binding portion (Fab) of the immunoglobulin molecule. Plotz and Oldstone presented evidence that autoimmunity can arise as a result of a cross-reaction between the idiotype on an antiviral antibody and a host cell receptor for the virus in question. In this case, the host-cell receptor is envisioned as an internal image of the virus, and the anti-idiotype antibodies can react with the host cells.
- Cytokine dysregulation – Cytokines have been recently divided into two groups according to the population of cells whose functions they promote: Helper T-cells type 1 or type 2. The second category of cytokines, which include IL-4, IL-10 and TGF-β (to name a few), seem to have a role in prevention of exaggeration of pro-inflammatory immune responses.
- Dendritic cell apoptosis – immune system cells called dendritic cells present antigens to active lymphocytes. Dendritic cells that are defective in apoptosis can lead to inappropriate systemic lymphocyte activation and consequent decline in self-tolerance.[24]
- Epitope spreading or epitope drift – when the immune reaction changes from targeting the primary epitope to also targeting other epitopes.[25] In contrast to molecular mimicry, the other epitopes need not be structurally similar to the primary one.
- Epitope modification or Cryptic epitope exposure – this mechanism of autoimmune disease is unique in that it does not result from a defect in the hematopoietic system. Instead, disease results from the exposure of cryptic N-glycan (polysaccharide) linkages common to lower eukaryotes and prokaryotes on the glycoproteins of mammalian non-hematopoietic cells and organs[26] This exposure of phylogenically primitive glycans activates one or more mammalian innate immune cell receptors to induce a chronic sterile inflammatory state. In the presence of chronic and inflammatory cell damage, the adaptive immune system is recruited and self–tolerance is lost with increased autoantibody production. In this form of the disease, the absence of lymphocytes can accelerate organ damage, and intravenous IgG administration can be therapeutic. Although this route to autoimmune disease may underlie various degenerative disease states, no diagnostics for this disease mechanism exist at present, and thus its role in human autoimmunity is currently unknown.
The roles of specialized immunoregulatory cell types, such as regulatory T cells, NKT cells, γδ T-cells in the pathogenesis of autoimmune disease are under investigation.
Classification
Autoimmune diseases can be broadly divided into systemic and organ-specific or localised autoimmune disorders, depending on the principal clinico-pathologic features of each disease.
- Systemic autoimmune diseases include coeliac disease, lupus erythematosus, Sjögren syndrome, sarcoidosis, scleroderma, rheumatoid arthritis, cryoglobulinemic vasculitis, and dermatomyositis. These conditions tend to be associated with autoantibodies to antigens which are not tissue specific. Thus although polymyositis is more or less tissue specific in presentation, it may be included in this group because the autoantigens are often ubiquitous t-RNA synthetases.
- Local syndromes which affect a specific organ or tissue:
- Endocrinologic: diabetes mellitus type 1, Hashimoto's thyroiditis, Addison's disease
- Gastrointestinal: Crohn's disease, pernicious anaemia
- Dermatologic: pemphigus vulgaris, vitiligo
- Haematologic: autoimmune haemolytic anaemia, idiopathic thrombocytopenic purpura
- Neurological: multiple sclerosis, myasthenia gravis, autoimmune encephalitis, gluten ataxia
Using the traditional "organ specific" and "non-organ specific" classification scheme, many diseases have been lumped together under the autoimmune disease umbrella. However, many chronic inflammatory human disorders lack the telltale associations of B and T cell driven immunopathology. In the last decade it has been firmly established that tissue "inflammation against self" does not necessarily rely on abnormal T and B cell responses.[27]
This has led to the recent proposal that the spectrum of autoimmunity should be viewed along an "immunological disease continuum," with classical autoimmune diseases at one extreme and diseases driven by the innate immune system at the other extreme. Within this scheme, the full spectrum of autoimmunity can be included. Many common human autoimmune diseases can be seen to have a substantial innate immune mediated immunopathology using this new scheme. This new classification scheme has implications for understanding disease mechanisms and for therapy development.[27]
Diagnosis
Diagnosis of autoimmune disorders largely rests on accurate history and physical examination of the patient, and high index of suspicion against a backdrop of certain abnormalities in routine laboratory tests (example, elevated C-reactive protein).
In several systemic disorders, serological assays which can detect specific autoantibodies can be employed. Localised disorders are best diagnosed by immunofluorescence of biopsy specimens.
Autoantibodies are used to diagnose many autoimmune diseases. The levels of autoantibodies are measured to determine the progress of the disease.
Treatments
Treatments for autoimmune disease have traditionally been immunosuppressive, anti-inflammatory, or palliative.[11] Managing inflammation is critical in autoimmune diseases.[28] Non-immunological therapies, such as hormone replacement in Hashimoto's thyroiditis or Type 1 diabetes mellitus treat outcomes of the autoaggressive response, thus these are palliative treatments. Dietary manipulation limits the severity of celiac disease. Steroidal or NSAID treatment limits inflammatory symptoms of many diseases. IVIG is used for CIDP and GBS. Specific immunomodulatory therapies, such as the TNFα antagonists (e.g. etanercept), the B cell depleting agent rituximab, the anti-IL-6 receptor tocilizumab and the costimulation blocker abatacept have been shown to be useful in treating RA. Some of these immunotherapies may be associated with increased risk of adverse effects, such as susceptibility to infection.
Helminthic therapy is an experimental approach that involves inoculation of the patient with specific parasitic intestinal nematodes (helminths). There are currently two closely related treatments available, inoculation with either Necator americanus, commonly known as hookworms, or Trichuris Suis Ova, commonly known as Pig Whipworm Eggs.[29][30][31][32][33]
T-cell vaccination is also being explored as a possible future therapy for autoimmune disorders.
Nutrition and autoimmunity
Vitamin D/Sunlight
- Because most human cells and tissues have receptors for vitamin D, including T and B cells, adequate levels of vitamin D can aid in the regulation of the immune system.[34] Vitamin D plays a role in immune function by acting on T cells and natural killer cells.[35] Research has demonstrated an association between low serum vitamin D and autoimmune diseases, including multiple sclerosis, type 1 diabetes, and Systemic Lupus Erythematosus (commonly referred to simply as lupus).[35][36][37] However, since photosensitivity occurs in lupus, patients are advised to avoid sunlight which may be responsible for vitamin D deficiency seen in this disease.[35][36][37] Polymorphisms in the vitamin D receptor gene are commonly found in people with autoimmune diseases, giving one potential mechanism for vitamin D's role in autoimmunity.[35][36] There is mixed evidence on the effect of vitamin D supplementation in type 1 diabetes, lupus, and multiple sclerosis.[35][36][37]
Omega-3 Fatty Acids
- Studies have shown that adequate consumption of omega-3 fatty acids counteracts the effects of arachidonic acids, which contribute to symptoms of autoimmune diseases. Human and animal trials suggest that omega-3 is an effective treatment modality for many cases of Rheumatoid Arthritis, Inflammatory Bowel Disease, Asthma, and Psoriasis.[38]
- While major depression is not necessarily an autoimmune disease, some of its physiological symptoms are inflammatory and autoimmune in nature. Omega-3 may inhibit production of interferon gamma and other cytokines which cause the physiological symptoms of depression. This may be due to the fact that an imbalance in omega-3 and omega-6 fatty acids, which have opposing effects, is instrumental in the etiology of major depression.[38]
Probiotics/Microflora
- Various types of bacteria and microflora present in fermented dairy products, especially Lactobacillus casei, have been shown to both stimulate immune response to tumors in mice and to regulate immune function, delaying or preventing the onset of nonobese diabetes. This is particularly true of the Shirota strain of L. casei (LcS). The LcS strain is mainly found in yogurt and similar products in Europe and Japan, and rarely elsewhere.[39]
- It has been theorized that free radicals contribute to the onset of type-1 diabetes in infants and young children, and therefore that the risk could be reduced by high intake of antioxidant substances during pregnancy. However, a study conducted in a hospital in Finland from 1997-2002 concluded that there was no statistically significant correlation between antioxidant intake and diabetes risk.[40] This study involved monitoring of food intake through questionnaires, and estimated antioxidant intake on this basis, rather than by exact measurements or use of supplements.
See also
- Autoimmune disease
- Protective autoimmunity
- Psychoneuroimmunology
References
- The Editors of Encyclopaedia Britannica (20 November 2018). "Autoimmunity". Health & Medicine. Encyclopædia Britannica. Archived from the original on 5 January 2021. Retrieved 5 January 2020.
{{cite web}}:|last=has generic name (help) - Delves, Peter J. (1998-01-01), "Autoimmunity", in Delves, Peter J. (ed.), Encyclopedia of Immunology (Second Edition), Oxford: Elsevier, pp. 292–296, ISBN 978-0-12-226765-9, retrieved 2021-01-06
- Erjavec SO, Gelfman S, Abdelaziz AR, Lee EY, Monga I, Alkelai A, Ionita-Laza I, Petukhova L, Christiano AM (Feb 2022). "Whole exome sequencing in Alopecia Areata identifies rare variants in KRT82". Nat Commun. 13 (1): 800. Bibcode:2022NatCo..13..800E. doi:10.1038/s41467-022-28343-3. PMC 8831607. PMID 35145093.
- Patt H, Bandgar T, Lila A, Shah N (2013). "Management issues with exogenous steroid therapy". Indian Journal of Endocrinology and Metabolism. 17 (Suppl 3): s612–s617. doi:10.4103/2230-8210.123548. PMC 4046616. PMID 24910822.
- Diamond, Betty; Lipsky, Peter E. (2014), Kasper, Dennis; Fauci, Anthony; Hauser, Stephen; Longo, Dan (eds.), "Autoimmunity and Autoimmune Diseases", Harrison's Principles of Internal Medicine (19 ed.), New York, NY: McGraw-Hill Education, archived from the original on 5 January 2021, retrieved 2021-01-05
- Arthur M. Silverstein: Autoimmunity: A History of the Early Struggle for Recognition, in: Ian R. Mackay, Noel R Rose: The Autoimmune Diseases (chapter 2), Academic Press, 2013
- Poletaev AB, Churilov LP, Stroev YI, Agapov MM (2012). "Immunophysiology versus immunopathology: Natural autoimmunity in human health and disease". Pathophysiology. 19 (3): 221–31. doi:10.1016/j.pathophys.2012.07.003. PMID 22884694.
- Stefanova I.; Dorfman J. R.; Germain R. N. (2002). "Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes". Nature. 420 (6914): 429–434. Bibcode:2002Natur.420..429S. doi:10.1038/nature01146. PMID 12459785. S2CID 993284.
- Pike B, Boyd A, Nossal G (1982). "Clonal anergy: the universally anergic B lymphocyte". Proceedings of the National Academy of Sciences. 79 (6): 2013–7. Bibcode:1982PNAS...79.2013P. doi:10.1073/pnas.79.6.2013. PMC 346112. PMID 6804951.
- Jerne N (1974). "Towards a network theory of the immune system". Annales d'Immunologie. 125C (1–2): 373–89. PMID 4142565.
- "Tolerance and Autoimmunity". Archived from the original on 2011-01-01. Retrieved 2007-03-19.
- Edwards JC, Cambridge G, Abrahams VM (1999). "Do self perpetuating B lymphocytes drive human autoimmune disease?". Immunology. 97 (2): 1868–1876. doi:10.1046/j.1365-2567.1999.00772.x. PMC 2326840. PMID 10447731.
- Grammatikos A, Tsokos G (2012). "Immunodeficiency and autoimmunity: lessons from systemic lupus erythematosus". Trends in Molecular Medicine. 18 (2): 101–108. doi:10.1016/j.molmed.2011.10.005. PMC 3278563. PMID 22177735.
- Klein J, Sato A (September 2000). "The HLA system. Second of two parts". New England Journal of Medicine. 343 (11): 782–6. doi:10.1056/NEJM200009143431106. PMID 10984567.
- Gregersen, Peter K.; Olsson, Lina M. (2009-01-01). "Recent Advances in the Genetics of Autoimmune Disease". Annual Review of Immunology. 27: 363–391. doi:10.1146/annurev.immunol.021908.132653. PMC 2992886. PMID 19302045.
- Women and Autoimmune Disorders By Krisha McCoy. Medically reviewed by Lindsey Marcellin, MD, MPH. Last Updated: 12/02/2009
- Ainsworth, Claire (Nov. 15, 2003). The Stranger Within Archived 2008-10-22 at the Wayback Machine. New Scientist (subscription). (reprinted here )
- Theory: High autoimmunity in females due to imbalanced X chromosome inactivation:
- Uz E, Loubiere LS, Gadi VK, et al. (June 2008). "Skewed X-chromosome Inactivation in Scleroderma". Clinical Reviews in Allergy & Immunology. 34 (3): 352–5. doi:10.1007/s12016-007-8044-z. PMC 2716291. PMID 18157513.
- Saunders K, Raine T, Cooke A, Lawrence C (2007). "Inhibition of Autoimmune Type 1 Diabetes by Gastrointestinal Helminth Infection". Infection and Immunity. 75 (1): 397–407. doi:10.1128/IAI.00664-06. PMC 1828378. PMID 17043101.
- "Parasite Infection May Benefit Multiple Sclerosis Patients". sciencedaily.com.
- Wållberg M, Harris R (2005). "Co-infection with Trypanosoma brucei brucei prevents experimental autoimmune encephalomyelitis in DBA/1 mice through induction of suppressor APCs". International Immunology. 17 (6): 721–8. doi:10.1093/intimm/dxh253. PMID 15899926.
- Edwards JC, Cambridge G (2006). "B-cell targeting in rheumatoid arthritis and other autoimmune diseases". Nature Reviews Immunology. 6 (5): 394–403. doi:10.1038/nri1838. PMID 16622478. S2CID 7235553.
- Kubach J, Becker C, Schmitt E, Steinbrink K, Huter E, Tuettenberg A, Jonuleit H (2005). "Dendritic cells: sentinels of immunity and tolerance". International Journal of Hematology. 81 (3): 197–203. doi:10.1532/IJH97.04165. PMID 15814330. S2CID 34998016.
- Induction of autoantibodies against tyrosinase-related proteins following DNA vaccination: Unexpected reactivity to a protein paralogue Archived May 3, 2008, at the Wayback Machine Roopa Srinivasan, Alan N. Houghton, and Jedd D. Wolchok
- Green R.S.; Stone E.L.; Tenno M.; Lehtonen E.; Farquhar M.G.; Marth J.D. (2007). "Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis". Immunity. 27 (2): 308–320. doi:10.1016/j.immuni.2007.06.008. PMID 17681821.
- McGonagle, D; McDermott, MF (Aug 2006). "A proposed classification of the immunological diseases". PLOS Medicine. 3 (8): e297. doi:10.1371/journal.pmed.0030297. PMC 1564298. PMID 16942393.
- Nikoopour E, Schwartz JA, Singh B (2008). "Therapeutic benefits of regulating inflammation in autoimmunity". Inflammation & Allergy - Drug Targets. 7 (3): 203–210. doi:10.2174/187152808785748155. PMID 18782028.
- Zaccone P, Fehervari Z, Phillips JM, Dunne DW, Cooke A (2006). "Parasitic worms and inflammatory diseases". Parasite Immunology. 28 (10): 515–23. doi:10.1111/j.1365-3024.2006.00879.x. PMC 1618732. PMID 16965287.
- Dunne DW, Cooke A (2005). "A worm's eye view of the immune system: consequences for evolution of human autoimmune disease". Nature Reviews Immunology. 5 (5): 420–6. doi:10.1038/nri1601. PMID 15864275. S2CID 24659866.
- Dittrich AM, Erbacher A, Specht S, et al. (2008). "Helminth Infection with Litomosoides sigmodontis Induces Regulatory T Cells and Inhibits Allergic Sensitization, Airway Inflammation, and Hyperreactivity in a Murine Asthma Model". The Journal of Immunology. 180 (3): 1792–9. doi:10.4049/jimmunol.180.3.1792. PMID 18209076.
- Wohlleben G, Trujillo C, Müller J, et al. (2004). "Helminth infection modulates the development of allergen-induced airway inflammation". International Immunology. 16 (4): 585–96. doi:10.1093/intimm/dxh062. PMID 15039389.
- Quinnell RJ, Bethony J, Pritchard DI (2004). "The immunoepidemiology of human hookworm infection". Parasite Immunology. 26 (11–12): 443–54. doi:10.1111/j.0141-9838.2004.00727.x. PMID 15771680. S2CID 32598886.
- Holick, Michael (December 2004). "Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease". The American Journal of Clinical Nutrition. 80 (6): 1678S–1688S. doi:10.1093/ajcn/80.6.1678S. PMID 15585788.
- Yang, Chen-Yen; Leung, Patrick S. C.; Adamopoulos, Iannis E.; Gershwin, M. Eric (2013-10-01). "The Implication of Vitamin D and Autoimmunity: a Comprehensive Review". Clinical Reviews in Allergy & Immunology. 45 (2): 217–226. doi:10.1007/s12016-013-8361-3. PMC 6047889. PMID 23359064.
- Dankers, Wendy; Colin, Edgar M.; Hamburg, Van; Piet, Jan; Lubberts, Erik (2017). "Vitamin D in Autoimmunity: Molecular Mechanisms and Therapeutic Potential". Frontiers in Immunology. 7: 697. doi:10.3389/fimmu.2016.00697. PMC 5247472. PMID 28163705.
- Agmon-Levin, Nancy; Theodor, Emanuel; Segal, Ramit Maoz; Shoenfeld, Yehuda (2013-10-01). "Vitamin D in Systemic and Organ-Specific Autoimmune Diseases". Clinical Reviews in Allergy & Immunology. 45 (2): 256–266. doi:10.1007/s12016-012-8342-y. PMID 23238772. S2CID 13265245.
- Simopoulos, Artemis (2002). "Omega-3 Fatty Acids in Inflammation and Autoimmune Diseases". Journal of the American College of Nutrition. 21 (6): 495–505. doi:10.1080/07315724.2002.10719248. PMID 12480795. S2CID 16733569.
- Matsuzaki, Takeshi; Akimitsu Takagi; Haruo Ikemura; Tetsuya Matsuguchi; Teruo Yokokura (March 2007). "Intestinal Microflora: Probiotics and Autoimmunity". The Journal of Nutrition. 137 (3): 798S–802S. doi:10.1093/jn/137.3.798S. PMID 17311978.
- Uusitalo, Liisa; Mike G Kenward; Suvi M Virtanen; Ulla Uusitalo; Jaakko Nevalainen; Sari Niinistö; Carina Kronberg-Kippilä; Marja-Leena Ovaskainen; Liisa Marjamäki; Olli Simell; Jorma Ilonen; Riitta Veijola; Mikael Knip (August 2008). "Intake of antioxidant vitamins and trace elements during pregnancy and risk of advanced beta cell autoimmunity in the child". The American Journal of Clinical Nutrition. 88 (2): 458–464. doi:10.1093/ajcn/88.2.458. PMID 18689383.
External links
- American Autoimmune Related Diseases Association: a nonprofit advocacy
- Immune Tolerance Network: a research-oriented resource
- Nobel Prize - The 1960 Nobel Prize in Physiology or Medicine was awarded to Frank M. Burnet and Peter B Medawar "for discovery of acquired immunological tolerance."
- The Immunology Database and Analysis Portal – an NIAID-funded database resource of reference and experiment data covering the entire immunology domain
- Understanding Autoimmune Diseases - US National Institute of Arthritis and Musculoskeletal and Skin Diseases