Cromatografía de gases
La cromatografía de gases es una técnica cromatográfica en la que la muestra se volatiliza y se inyecta en la cabeza de un mechero de una columna cromatográfica. La elución se produce por el flujo de una fase móvil de gas inerte. A diferencia de los otros tipos de cromatografía, la fase móvil no interactúa con las moléculas del analito; su única función es la de transportar el analito a través de la columna.

Existen dos tipos de cromatografía de gases (GC): la cromatografía gas-sólido (GSC) y la cromatografía gas-líquido (GLC), siendo esta última la que se utiliza más ampliamente, y que se puede llamar simplemente cromatografía de gases (GC). En la GSC la fase estacionaria es sólida y la retención de los analitos en ella se produce mediante el proceso de adsorción. Precisamente este proceso de adsorción, que no es lineal, es el que ha provocado que este tipo de cromatografía tenga aplicación limitada, ya que la retención del analito sobre la superficie es semipermanente y se obtienen picos de elución con colas. Su única aplicación es la separación de especies gaseosas de bajo peso molecular. La GLC utiliza como fase estacionaria moléculas de líquido inmovilizadas sobre la superficie de un sólido inerte.
La GC se lleva a cabo en un cromatógrafo de gases. Este consta de diversos componentes como el gas portador, el sistema de inyección de muestra, la columna (generalmente dentro de un horno), y el detector.
Historia
La cromatografía data de 1903 en la obra del científico ruso, Mijaíl Tsvet. El estudiante de doctorado alemán Fritz Prior desarrolló la cromatografía de gas de estado sólido en 1947. Archer John Porter Martin, que fue galardonado con el Premio Nobel por su trabajo en el desarrollo de la cromatografía líquido-líquido (1941) y de papel (1944), sentó las bases para el desarrollo de la cromatografía de gas y más tarde de la cromatografía líquido-gas (1950). Erika Cremer sentó las bases y supervisó gran parte del trabajo de Prior.
Gas portador
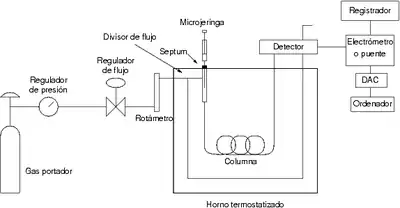
El gas portador cumple básicamente dos propósitos: Transportar los componentes de la muestra, y crear una matriz adecuada para el detector. Un gas portador debe reunir ciertas condiciones:
- Debe ser inerte para evitar interacciones (tanto con la muestra como con la fase estacionaria)
- Debe ser capaz de minimizar la difusión gaseosa
- Fácilmente disponible y puro
- Económico
- Adecuado al detector a utilizar...
El gas portador debe ser un gas inerte, para impedir su reacción con el analito o la columna. Generalmente se emplean gases como el helio, argón, nitrógeno, hidrógeno o dióxido de carbono, y la elección de este gas en ocasiones depende del tipo de detector empleado. El almacenaje del gas puede ser en balas normales o empleando un generador, especialmente en el caso del nitrógeno y del hidrógeno. Luego tenemos un sistema de manómetros y reguladores de flujo para garantizar un flujo estable y un sistema de deshidratación del gas, como puede ser un tamiz molecular.
Generalmente la regulación de la presión se hace a dos niveles: un primer manómetro se sitúa a la salida de la bala o generador del gas y el otro a la entrada del cromatógrafo, donde se regula el flujo. Las presiones de entrada varían entre 10 y 25 psi, lo que da lugar a caudales de 25 a 150 mL/min en columnas de relleno y de 1 a 25 mL/min en columnas capilares. Para comprobar el caudal se puede utilizar un rotámetro o un simple medidor de pompas de jabón, el cual da una medida muy exacta del caudal volumétrico que entra a la columna.
La pureza de los gases es sumamente importante, se requieren niveles 4.5 o mayores es decir 99.995 % de pureza. Sin embargo, debido al cuidado que se debe tener con la fase activa de la columna, se hace completamente necesario la instalación de trampas a la entrada del gas portador, estas trampas obviamente tienen una capacidad limitada, pero son importantísimas al momento de usar el cromatógrafo. Estas trampas evitan el ingreso de hidrocarburos, agua y CO entre otros.
Sistema de inyección de muestra
La inyección de muestra es un apartado crítico, ya que se debe inyectar una cantidad adecuada, y debe introducirse de tal forma (como un "tapón de vapor") que sea rápida para evitar el ensanchamiento de las bandas de salida; este efecto se da con cantidades elevadas de analito. El método más utilizado emplea una microjeringa (de capacidades de varios microlitros) para introducir el analito en una cámara de vaporización instantánea. Esta cámara está a 50 °C por encima del punto de ebullición del componente menos volátil, y está sellada por una junta de goma de silicona septa o septum.
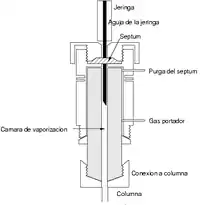
Si es necesaria una reproducibilidad del tamaño de muestra inyectado se puede usar una válvula de seis vías o válvula de inyección, donde la cantidad a inyectar es constante y determinada por el tamaño del bucle de dicha válvula.

Si la columna empleada es rellena, el volumen a inyectar será de unos 20 μL, y en el caso de las columnas capilares dicha cantidad es menor, de 1 μL, y según el tipo de columna capilar (ya que existen columnas de distinto diámetro interno) es que si se utiliza todo el volumen de muestra inyectado. Para obtener menor cantidad de volumen, se utiliza un divisor de flujo (la inyección se conoce como modo "split") a la entrada de la columna que desecha parte del analito introducido. Si se utiliza todo el volumen de muestra la inyección es de tipo "splitless". El modo splitless se empleó más para determinar cantidades pequeñas o trazas (determinaciones ambientales).
Si se inyecta 1 microlitro de solvente -por ejemplo agua-, al pasar a la fase vapor su volumen se multiplicará por mil. Es decir, un microlitro de agua pasaría a ser 1 mL de agua en gas; como el volumen del puerto de inyección es limitado, se emplean split pulsado u otras configuraciones para garantizar el ingreso adecuado de las muestras.
En caso de muestras sólidas, simplemente se introducen en forma de disolución, ya que en la cámara de vaporización instantánea el disolvente se pierde en la corriente de purga y no interfiere en la elución.
Según las curvas de Van Demter (HEPT vs. Velocidad Lineal), el mejor gas a usar en la columna cromatográfica como portador de los analitos es el hidrógeno, sin embargo dada su peligrosidad, es más usado como gas de encendido en el detector FID, junto con el aire. Luego vienen, respectivamente, helio y nitrógeno.
El gas hidrógeno es el mejor portador y los flujos que manejan los cromatógrafos no son peligrosos, además a la salida de estos generalmente existen restrictores de llama que evitan la propagación de un posible incendio. Se puede recomendar el uso de hidrógeno debido a, primero por su bajo precio respecto a los otros gases y por la resolución de los picos que se muestran en los cromatogramas.
La relación para la ignición entre hidrógeno y aire es de 4,1% para el límite inferior y del 74,8% para el superior a 101,3Kpa y 298K (Safety Standard for Hydrogen and Hydrogen Systems, NASA), y se tiene que estar en presencia de una chispa o zona de calentamiento alta (desde 520 °C).
Columnas y sistemas de control de temperatura
En GC se emplean dos tipos de columnas: las empacadas o de relleno y las tubulares abiertas o capilares. Estas últimas son más comunes en la actualidad (2005) debido a su mayor rapidez y eficiencia. La longitud de estas columnas es variable, de 2 a 60 metros, y están construidas de acero inoxidable, vidrio, sílice fundida o teflón. Debido a su longitud y a la necesidad de introducirlas en un horno, las columnas suelen enrollarse en una forma helicoidal con longitudes de 10 a 30 cm, dependiendo del tamaño del horno.
La temperatura es una variable importante, ya que de ella va a depender el grado de separación de los diferentes analitos. Para ello, debe ajustarse con una precisión de décimas de grado. Dicha temperatura depende del punto de ebullición del analito o analitos, como también la máxima temperatura de funcionamiento de la columna (fase estacionaria), y por lo general se ajusta a un valor igual o ligeramente superior a él. Para estos valores, el tiempo de elución va a oscilar entre 2 y 30-40 minutos. Si tenemos varios componentes con diferentes puntos de ebullición, se ajusta la llamada rampa de temperatura con lo cual ésta va aumentando ya sea de forma continua o por etapas. En muchas ocasiones, el ajustar correctamente la rampa puede significar separar bien o no los diferentes analitos. Es recomendable utilizar temperaturas bajas para la elución, ya que -aunque a mayor temperatura la elución es más rápida- se corre el riesgo de descomponer el analito. Se puede programar la rampa tanto para aumentar como para disminuir la temperatura del horno para que no haya solapamiento de los picos.
Detectores
El detector es la parte del cromatógrafo que se encarga de determinar cuándo ha salido el analito por el final de la columna. Las características de un detector ideal son:
- Sensibilidad: Es necesario que pueda determinar con precisión cuándo sale analito y cuando sale sólo el gas portador. Tienen sensibilidades entre 10-8 y 10-15 g/s de analito.
- Respuesta lineal al analito con un rango de varios órdenes de magnitud.
- Tiempo de respuesta corto, independiente del caudal de salida.
- Intervalo de temperatura de trabajo amplio, por ejemplo desde temperatura ambiente hasta unos 350-400 °C, temperaturas típicas trabajo.
- Estabilidad y reproducibilidad, es decir, a cantidades iguales de analito debe dar salidas de señal iguales.
- Alta fiabilidad y manejo sencillo, o a prueba de operadores inexpertos.
- Respuesta semejante para todos los analitos, o
- Respuesta selectiva y altamente predecible para un reducido número de analitos.
Algunos tipos de detectores:
- Detector de ionización de llama (FID, Flame Ionization Detector).
- Detector de conductividad térmica (TCD, Thermical Conductivity Detector).
- Detector termoiónico (TID, ThermoIonic Detector).
- Detector de captura de electrones (ECD, Electrón-Capture Detector).
- Detector de emisión atómica (AED, Atomic Emission Detector).
-_ANTOFAGASTA-1_143.jpg.webp)
Otros detectores minoritarios son el detector fotométrico de llama (PFD), empleado en compuestos como pesticidas e hidrocarburos que contengan fósforo o azufre. En este detector se hace pasar el gas eluido por una llama hidrógeno/oxígeno donde parte del fósforo se convierte en una especie HPO, la cual emite a λ = 510 y 526 nm, y simultáneamente el azufre se convierte en S2, con emisión a λ = 394 nm. Dicha radiación emitida se detecta con un fotómetro adecuado. Se han podido detectar otros elementos, como algunos halógenos, nitrógeno, estaño, germanio y otros.
En el detector de fotoionización (PID), el gas eluido al final de la columna se somete a una radiación ultravioleta con energías entre 8,3 y 11,7 eV, correspondiente a una λ = 106-149 nm. Mediante la aplicación de un potencial a la celda de ionización se genera una corriente de iones, la cual es amplificada y registrada.
Columnas y tipos de fases estacionarias
- Columnas de relleno
Las columnas de relleno o empacadas consisten en unos tubos de vidrio, metal (inerte a ser posible como el acero inoxidable, níquel, cobre o aluminio) o teflón, de longitud de 2 a 3 metros y un diámetro interno de unos pocos milímetros, típicamente de 2 a 4. El interior se rellena con un material sólido, finamente dividido para tener una máxima superficie de interacción y recubierto con una capa de espesores entre 50 nm y 1 μm. Para que puedan introducirse en el horno, se enrollan convenientemente.
El material de relleno ideal consiste en pequeñas partículas, esféricas y uniformes, con una buena resistencia mecánica, para tener una máxima superficie donde interaccionar la fase estacionaria y el analito. La superficie específica mínima ha de ser de 1 m²/g. Como todos los componentes de columnas para GC, debe ser inerte a altas temperaturas (~400 °C) y humectarse uniformemente con la fase líquida estacionaria durante el proceso de fabricación. El material preferido actualmente (2005) es la tierra de diatomeas natural, debido a su tamaño de poro natural. Estas especies, ya extinguidas, utilizaban un sistema de difusión molecular para tomar nutrientes del medio y expulsar sus residuos. Por lo tanto son materiales especialmente útiles, debido a que el sistema de absorción superficial del analito y la fase estacionaria es parecido.
El tamaño es crítico a la hora de darse el proceso de interacción del analito, y a menores tamaños la eficacia de la columna es mejor. Pero existe el problema de la presión necesaria para hacer circular un caudal estable de gas portador por la columna, ya que dicha presión es inversamente proporcional al cuadrado del diámetro de dichas partículas. Así, el tamaño mínimo para usar presiones máximas de 50 psi es de 250 a 149 μm.
- Columnas capilares
Las columnas capilares son de dos tipos básicos: las de pared recubierta (WCOT) y las de soporte recubierto (SCOT). Las WCOT son simplemente tubos capilares donde la pared interna se ha recubierto con una finísima capa de fase estacionaria. Las columnas SCOT tienen en su parte interna una fina capa de material absorbente como el empleado en las columnas de relleno (tierra de diatomeas) donde se ha adherido la fase estacionaria. Las ventajas de las WCOT frente a las SCOT es la mayor capacidad de carga, ya que en su fabricación se emplean mayores cantidades de fase estacionaria, al ser la superficie de intercambio mayor. Por orden de eficacia, en primer lugar están las WCOT, luego las SCOT y por último las columnas de relleno.
Las columnas WCOT se fabrican a partir de sílice fundida, conocidas como columnas tubulares abiertas de sílice fundida o FSOT. Estas columnas se fabrican a partir de sílice especialmente pura, sin apenas contenido de óxidos metálicos. Debido a la fragilidad inherente a este material, en el mismo proceso de obtención del tubo se recubre con una capa de poliimida, de esta forma la columna puede enrollarse con un diámetro de unos pocos centímetros. Estas columnas, con propiedades como baja reactividad, resistencia física y flexibilidad, han sustituido a las WCOT clásicas.
Las columnas FSOT tienen diámetros internos variables, entre 250 y 320 μm (para columnas normales) y 150-200 μm para columnas de alta resolución. Estas últimas requieren menor cantidad de analito y un detector más sensible, al eluir menor cantidad de gas. Existen asimismo columnas macrocapilares con diámetros de hasta 530 μm, que admiten cantidades de analito comparables a las de relleno pero con mejores prestaciones.
En estas columnas existe un problema debido a la adsorción del analito sobre la superficie de la sílice fundida, adsorción debida a la presencia de grupos silanol (Si-OH), los cuales interaccionan fuertemente con moléculas polares orgánicas. Se suele solventar este inconveniente inactivando la superficie por sililación con dimetilclorosilano (DMCS). La adsorción debida a los óxidos metálicos se ve paliada en gran parte por la elevada pureza de la sílice empleada.
- La fase estacionaria
Las propiedades necesarias para una fase estacionaria líquida inmovilizada son:
- Características de reparto (factor de capacidad κ' y factor de selectividad α) adecuados al analito.
- Baja volatilidad, el punto de ebullición de la fase estacionaria debe ser al menos 100 °C mayor que la máxima temperatura alcanzada en el horno.
- Baja reactividad.
- Estabilidad térmica, para evitar su descomposición durante la elución.
Existen como mucho una docena de disolventes con estas características. Para elegir uno, debe tenerse en cuenta la polaridad del analito, ya que a mayor polaridad del analito, mayor polaridad deberá tener la fase estacionaria. Algunas fases estacionarias utilizadas actualmente (2005) son:
- Polidimetilsiloxano, fase no polar de uso general para hidrocarburos, aromáticos, polinucleares, drogas, esteroides y PCB.
- Poli(fenilmetildifenil)siloxano (10% fenilo), para ésteres metílicos de ácidos grasos, alcaloides, drogas y compuestos halogenados.
- Poli(fenilmetil)siloxano (50% fenilo), para drogas, esteroides, pesticidas y glicoles.
- Poli(trifluoropropildimetil)siloxano, para aromáticos clorados, nitroaromáticos, bencenos alquilsustituidos.
- Polietilenglicol,sirve para compuestos polares, también para compuestos como glicoles, alcoholes, éteres, aceites esenciales.
- Poli(dicianoalildimetil)siloxano, para ácidos grasos poliinsaturados, ácidos libres y alcoholes.
Generalmente, en columnas comerciales, la fase estacionaria se presenta enlazada y entrecruzada para impedir su pérdida durante las operaciones de elución o lavado. De esta forma se obtiene una monocapa adherida químicamente a la superficie de la columna. La reacción implicada suele ser la adición de un peróxido al líquido a fijar, iniciándose una reacción por radicales libres que tiene como resultado la formación de un enlace carbono-carbono que además incrementa su estabilidad térmica. Otra forma es la irradiación con rayos gamma.
Otro tipo de fase estacionaria son las quirales, lo cual permite resolver mezclas enantioméricas. Este tipo de fases suelen ser aminoácidos quirales o algún derivado adaptado al trabajo en columna.
El grosor de la película varía entre 0,1 y 5 μm; el grosor depende de la volatilidad del analito. Así, un analito muy volátil requerirá una capa gruesa para aumentar el tiempo de interacción y separar más efectivamente los diferentes componentes de la mezcla. Para columnas típicas (diámetros internos de 0,25 o 0,32 mm) se emplean grosores de 0,25 μm, y en las columnas macrocapilares el grosor sube hasta 1 μm. El grosor máximo suele ser de 8 μm
Aplicaciones
La GC tiene dos campos de aplicación importantes. Por una parte su capacidad para separar mezclas orgánicas complejas, compuestos organometálicos y sistemas bioquímicos. Su otra aplicación es como método para determinar cuantitativa y cualitativamente los componentes de la muestra. Para el análisis cualitativo se suele emplear el tiempo de retención, que es único de cada compuesto en condiciones determinadas (mismo gas portador, rampa de temperatura y flujo), o el volumen de retención. En aplicaciones cuantitativas, integrando las áreas de cada compuesto o midiendo su altura, con los calibrados adecuados, se obtiene la concentración o cantidad presente de cada analito.
Montaje de técnicas
El montaje de una técnica analítica de CG es netamente empírico, el perfil de los analitos que se quiera determinar, la elección de la fase móvil, los tiempos de retención (elución) estarán dados exclusivamente por las condiciones particulares de la columna (fase estacionaria) frente al equipo. Las rampas de temperatura a seleccionar bien pueden isotérmicas o escalonadas.
La elección del gás dependerá del tipo de detector, la elección de la columna (fase estacionaria) dependerá de la polaridad de los compuestos a separar, el detector dependerá del tipo de compuestos a detectar.
Usualmente una técnica analítica de GC consumirá muchas horas de un cromatografista en ser desarrollada e instalada por el método del ensayo y error antes de ser validada como real.
La elección de los estándares es fundamental en el desarrollo de la técnica. La estabilización de la línea base de la fase móvil en la fase estacionaria ( posterior al frente del solvente) a través del tiempo es fundamental para establecer un método. Una línea de base (solvente) poco estable o irregular que cambia de intensidad frente al detector a medida que eluye debe ser afinada y estabilizada antes de introducir los analitos.
El layout de los parámetros del rango de temperatura del horno, la adecuada elección de la columna y su fase estacionaria (incluye, tipo, largo y diámetro), la elección adecuada del tipo de detector, las temperaturas del detector e inyector, los volúmenes de analito, deberán ser establecidas de modo tal que se obtenga la mayor eficacia en separar los analitos, y con la mejor resolución posible. La pureza de la muestra dependerá de la preparación previa de la misma.
La CG es una metodología altamente efectiva y su performance permite una amplia gama de posibilidades para la química analítica en compuestos orgánicos. Una derivación de esta técnica es la Cromatografía HPLC que funciona sobre la base de la afinidad del analito por la fase móvil líquida en vez de gaseosa.
La sensibilidad de la técnica GC puede incluso detectar microgramos del analito si está bien montada. La cuantificación se basa en cálculos del área bajo la curva que es proporcional a la concentración del analito. Comúnmente se usa en estándar interno de trabajo.
Véase también
- elución (en inglés)
Bibliografía
- Skoog, Douglas A. y Leary, James J. (1994). Análisis Instrumental. Armenia: McGraw-Hill. ISBN 84-481-0191-X.
- McNair, Harold M. & Miller, James M. (1998). Basic Gas Chromatography. Canada: John Wiley & Sons, Inc. ISBN 0-471-17260-X (alk. paper); ISBN 0-471-17261-8 (pbk.: alk. paper).
| Control de autoridades |
|
|---|
 Datos: Q677065
Datos: Q677065 Multimedia: Gas chromatography / Q677065
Multimedia: Gas chromatography / Q677065