Enfermedad de Wilson
La enfermedad de Wilson o degeneración hepatolenticular es una enfermedad hereditaria autosómica recesiva, con una incidencia de alrededor de 1/50 000.[4] Su hecho principal es la acumulación de cobre en los tejidos, manifestada por síntomas neurológicos (dificultad de coordinación, temblores), también catarata y enfermedades hepáticas (cirrosis, insuficiencias hepáticas). La enfermedad afecta por igual a hombres y a mujeres y se ha descrito en todas las razas.
| Enfermedad de Wilson | ||
|---|---|---|
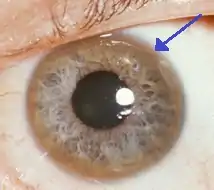 El anillo de Kayser-Fleischer (franja marrón en el borde del iris) es frecuente en la enfermedad de Wilson, especialmente cuando los síntomas neurológicos están presentes. | ||
| Especialidad | endocrinología | |
| Síntomas | Hinchazón de las piernas, piel amarillenta, cambios de personalidad[1] | |
| Causas | genéticas | |
| Diagnóstico diferencial | Hepatopatía crónica, enfermedad de Parkinson, esclerosis múltiple, temblor esencial[2] | |
| Sinónimos | ||
| Degeneración hepatolenticular[3] | ||
Esta anomalía genética transmitida como rasgo autosómico recesivo se caracteriza por un bajo nivel sérico de cobre y de ceruloplasmina (por el daño hepático, no por la propia enfermedad), mientras que aumenta el cobre en orina y en los tejidos. De esta manera aparecen grandes cantidades de cobre en el hígado, el bazo, el cerebro, la córnea y el riñón, los que pueden producir espasmos musculares, ataxia, atetosis, babeo, osteoporosis, convulsiones, disfagia, disartria y en algunos casos dolor de cabeza y ansiedad.[5]
A menos que se establezca un tratamiento la enfermedad causa la muerte precozmente. Es probable que el gen defectuoso responsable sea ATP7B, que codifica un transportador de cobre (también una ATPasa tipo P) que transfiere el metal desde los hepatocitos hasta la bilis, situado en el brazo largo del cromosoma 13.[6]

- Hipertransaminasemia sin etiología filiada
- Postura anormal de brazos y piernas
- Confusión o delirio
- Demencia
- Dificultad y rigidez para mover los brazos y las piernas
- Dificultad para caminar (ataxia)
- Cambios emocionales o conductuales
- Agrandamiento del abdomen (distensión abdominal)
- Cambios de personalidad
- Pérdida del deseo sexual
- Fobias, angustia (neurosis)
- Movimientos lentos
- Lentitud o disminución de los movimientos y expresiones faciales
- Deterioro del lenguaje
- Anillo de Kayser-Fleischer (anillo de color dorado oscuro o marrón que rodea el iris del ojo por depósito de cobre en la membrana de Descemet)
- Esplenomegalia
- Temblores en los brazos o en las manos
- Movimientos incontrolables
- Movimientos impredecibles o espasmódicos
- Vómito con sangre
- Debilidad
- Piel amarilla o color amarillo de la esclerótica del ojo (ictericia)
- Uñas azuladas
Diagnóstico
- Movimiento ocular limitado.
- Anillos de color marrón o rojizo alrededor del iris (anillo de Kayser-Fleischer).
- Conteo sanguíneo completo (CSC).
- Ceruloplasmina en suero.
- Cobre en suero.
- Ácido úrico en suero.
- Cobre en orina.
- Radiografía abdominal.
- Imágenes por resonancia magnética del abdomen.
- Tomografía computarizada del abdomen.
- Tomografía computarizada de la cabeza.
- Imágenes por resonancia magnética de la cabeza.
- Biopsia del hígado.
- Examen ocular.
Tratamiento
El objetivo del tratamiento es reducir la cantidad de cobre en los tejidos. Esto se hace mediante un procedimiento llamado quelación, en donde ciertos medicamentos se pueden fijar al cobre y ayudar eliminarlo a través de los riñones o los intestinos. El tratamiento debe hacerse de por vida.
Se pueden utilizar los siguientes medicamentos:
- La penicilamina (Cuprimine, Depen) se fija al cobre y lleva a un aumento en la eliminación de este elemento a través de la orina. Hay que procurar la ingesta de Vitamina B6 con la d-penicilamina.
- La trientina (Syprine) se fija (quela) al cobre e incrementa su excreción a través de la orina.
- El acetato de zinc (Galzin) bloquea la absorción (ingreso) del cobre en el tracto intestinal.
También se pueden utilizar los suplementos de vitamina E.
Algunas veces, los medicamentos que quelan el cobre, especialmente la penicilamina, pueden afectar el funcionamiento del cerebro y del sistema nervioso (función neurológica). Existen otros medicamentos bajo investigación que se fijan al cobre sin afectar la función neurológica.
Dieta
Además de los medicamentos adecuados, se debe aplicar la dieta correcta para el tratamiento de esta enfermedad. Esta consiste fundamentalmente en controlar la ingestión de alimentos que contengan cobre.
La lista siguiente contiene una referencia de alimentos ricos en este mineral que los enfermos de Wilson deben evitar.
Alimentos ricos en cobre: chocolate y todos sus derivados, vísceras en general (hígado, riñón, mollejas, corazón, sesos) todos los patés, frutos secos (nueces, almendras, maní, etc.), frutas deshidratadas, brocoli o brecol, verduras oscuras (se desaconsejan los caldos con muchas verduras), pescados, mariscos, champiñones, entre otros.
Alimentos que no contienen cobre: arroz y pollo.
Los complementos de vitamina E pueden ser interesantes para combatir los daños que la penicilamina pueda ocasionar en el hígado; así también se recomienda suministrar algún complemento vitamínico rico en vitamina B6.
Se debe abandonar por completo el consumo de alcohol.
También queda prohibido fumar.
No debe beberse agua mineral que contenga más de 100 mg de cobre por litro. El agua de grifo tampoco resulta muy conveniente porque contiene grandes cantidades de cobre por disolución del mismo de las rocas y conducciones de agua. Mejor beber agua desmineralizada.
Referencias
- «Wilson Disease». web.archive.org. 4 de octubre de 2016. Archivado desde el original el 4 de octubre de 2016. Consultado el 16 de noviembre de 2022.
- Lynn, D. Joanne; Newton, Herbert B.; Rae-Grant, Alexander (2004). The 5-minute Neurology Consult (en inglés). Lippincott Williams & Wilkins. ISBN 978-0-683-30723-8. Consultado el 16 de noviembre de 2022.
- «Medlineplus: Enfermedad de Wilson». Consultado el 22 de noviembre de 2013. «Nombres alternativos: Degeneración hepatolenticular ».
- Rodriguez-Castro, Kryssia Isabel; Hevia-Urrutia, Francisco Javier; Sturniolo, Giacomo (2015). «Wilson’s disease: A review of what we have learned». World Journal of Hepatology 7 (29): 2859-2870. doi:10.4254/WJH.v7.i29.2859. Consultado el 1 de diciembre de 2016.
- (2011 TEMIS medical S.L Barcelona)
- Medlineplus: Enfermedad de Wilson. Publicado el 8-7-2008. Consultado el 20-1-2010
Enlaces externos
- Enfermedad del Wilson desde el punto de vista de un paciente
- Asociación Española de Familiares y Enfermos de Wilson
- Asociación Costarricense de Pacientes con Enfermedad de Wilson
- Información general sobre la enfermedad de Wilson
| Control de autoridades |
|
|---|
 Datos: Q117121
Datos: Q117121 Multimedia: Wilson's disease / Q117121
Multimedia: Wilson's disease / Q117121