NF-κB
El NF-kB (factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas) es un complejo proteico que controla la transcripción del ADN. NF-kB se encuentra en la mayoría de tipos de células animales y está implicado en la respuesta celular frente a estímulos como el estrés, las citoquinas, radiación ultravioleta, LDL oxidadas y antígenos bacterianos o virales.[1][2][3][5][6] El NF-κB juega un papel clave en la regulación de la respuesta inmune debida a la infección (las cadenas ligeras kappa son componentes cruciales de las inmunoglobulinas). La regulación defectuosa del NF-kB está relacionada con el cáncer, enfermedades inflamatorias y autoinmunes, shock séptico, infecciones virales o un desarrollo inmune inadecuado. También está implicado en procesos de plasticidad sináptica y de memoria.[7][8][9][10][11]
Descubrimiento
El NF-κB fue descubierto en el laboratorio por el galardonado con el Premio Nobel David Baltimore a partir de su interacción con una secuencia de 11 pares de bases en la región potenciadora (enhancer) de la cadena ligera kappa de la inmunoglobulina en células B.[12]
Estructura
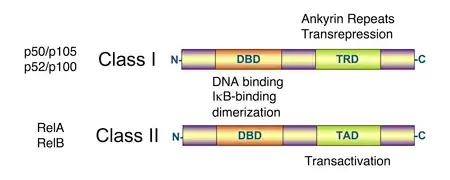
Todas las proteínas de la familia de los NF-κB comparten un dominio de homología Rel en su extremo N-terminal. Una subfamilia de proteínas NF-κB, incluidas RelA, RelB y c-Rel, tienen un dominio de transactivación en su extremo C-terminal. En contraste, las proteínas NF-κB1 y NF-κB2 son sintetizadas como precursores, p105 y p100, que tras madurar dan lugar a las subunidades de NF-κB, p50 y p52 respectivamente. La maduración de p105 y p100 está mediada por la vía ubiquitina/proteosoma e implica la degradación selectiva del C-terminal de la región que contiene repeticiones de anquirina. Mientras que la formación de p52 a partir de p100 es un proceso altamente regulado, la p50 se forma por un proceso constitutivo de la p105.[13][14]
Miembros
Los miembros de la familia del NF-κB comparten estructura homóloga con la oncoproteína retroviral v-Rel, lo que da lugar a su clasificación como proteínas NF-κB/Rel.[1]
En mamíferos existen 5 miembros de la familia NF-κB:[15]
| Clase | Proteína | Alias | Gen |
|---|---|---|---|
| I | NF-κB1 | p105 → p50 | NFKB1 |
| NF-κB2 | p100 → p52 | NFKB2 | |
| II | RelA | p65 | RELA |
| RelB | RELB | ||
| c-Rel | REL |
Distribución de especies y evolución
Además de en mamíferos, el NF-κB se encuentra también en animales más sencillos.[16] Entre ellos los cnidarios (como las anémonas de mar, el coral o la hidra), poríferos (esponjas), el parásito unicelular eucariota Capsaspora owczarzaki e insectos (como polillas, mosquitos y moscas de la fruta). La sucesión de los genomas de los mosquitos A. aegypti y A. gambiae, y la mosca de la fruta D. melanogaster ha permitido realizar estudios comparativos genéticos y evolutivos de NF-κB. En estas especies de insectos, la activación de NF-κB está provocada por la vía Toll (que evolucionó de forma independiente en insectos y mamíferos) y por la vía IMD (inmunodeficiencia).[17]
Señalización
Activación
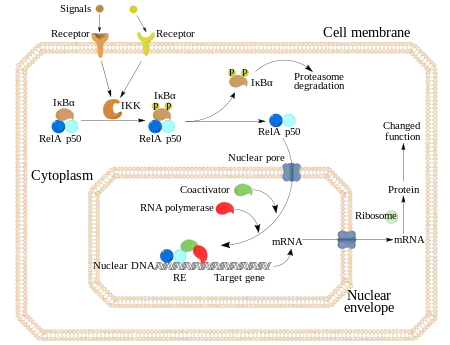
El NF-κB es relevante en la regulación de la respuesta celular ya que pertenece a la categoría de los factores de transcripción primarios de “acción rápida”, como son los factores de transcripción que están presentes en las células en un estado de inactivación y que no requieren una nueva síntesis de proteínas para ser activados (otros miembros de esta familia incluyen factores de transcripción tales como c-Jun, STATs, y receptores nucleares hormonales. Esto permite al NF-κB ser la primera respuesta a estímulos celulares nocivos. Los inductores de la actividad del NF-κB son altamente variables, y pueden ser desde especies reactivas de oxígeno (ROS), factores de necrosis tumoral alfa (TNF α), interleucina 1-beta (IL-1β), lipopolisacáridos bacteriales (LPS), isoproterenol, cocaína e incluso radiaciones iónicas.[19]
El receptor activador del factor nuclear kappa B (RANK), que es un tipo de TNFR, es un activador central del NF-κB. La osteoprotegerina (OPG), que es un receptor señuelo homólogo al ligando de RANK, inhibe RANK uniéndose a RANKL, y así, la osteoprotegerina está estrechamente relacionada con la regulación de la activación de NF-κB.[20]
Muchos productos bacteriales así como la estimulación de una gran variedad de receptores de la superficie celular inducen la activación de NF-κB así como también cambios rápidos en la expresión génica.[1] La identificación de receptores de tipo Toll (TLRs) como patrones específicos de reconocimiento molecular, y el descubrimiento que la estimulación de TLRs conduce a la activación de NF-κB, mejoró la comprensión de como los diferentes patógenos pueden llegar a activar NF-κB. Por ejemplo, diversos estudios han identificado TLR4 como un receptor para el componente LPS de las bacterias Gram-Negativas.[21] Los TLRs son reguladores esenciales tanto de las respuestas inmunes innatas como de las adaptativas.[22]
A diferencia de RelA, RelB, y c-Rel, las subunidades p50 y p52 de NF-κB no contienen dominios de transactivación en sus mitades C-terminales. No obstante, las p50 y p52 de NF-κB juegan un papel importante modulando la especificidad de la función de NF-κ . Aunque los homodímeros de p50 y p52 son, en general, represores del lugar de transcripción de κB, ambos (p50 y p52) participan en la transactivación de genes diana formando heterodímeros con RelA, RelB, o c-Rel.[23] Además, los homodímeros p50 y p52 también se unen a la proteína nuclear Bcl-3, pudiendo dichos complejos funcionar como activadores transcripcionales.[24][25][26]
Inhibición
En células no estimuladas, los dímeros de NF-κB son secuestrados en el citoplasma por una familia de inhibidores, llamados IκBs (Inhibidores de κB), los cuales son proteínas que contienen múltiples copias de una secuencia llamada “repeticiones de anquirina”. En virtud de estos dominios, las proteínas IκB enmascaran la secuencia de localización nuclear (NLS) de las proteínas NF-κB y las mantienen secuestradas en un estado de inactivación en el citoplasma.[27]
Las IκBs forman una familia de proteínas relacionadas, que tienen un dominio N-terminal regulador, seguido por seis o más repeticiones de anquirina y un dominio PEST cerca de su C-terminal. Aunque la familia de las IκB consiste en IκBα, IκBβ, IκBε, y Bcl-3, la principal y más estudiada IκB es la IκBα. Debido a la presencia de repeticiones de anquirina en las mitades C-terminales, p105 y p100 también funcionan como proteínas IκB. La mitad C-terminal de p100, que a menudo es conocida como IκBδ, también funciona como inhibidor.[28][29] La degradación de IκBδ en respuesta a estímulos de desarrollo, tales como aquellos transducidos a través de LTβR, potencia la activación del dímero NF-κB a través de una vía NIK dependiente no canónica.[28][30]
La activación de NF-κB se inicia a través de la degradación inducida por señal de proteínas IκB. Esto ocurre principalmente mediante la activación de una quinasa llamada IκB quinasa (IKK). La IKK está formada por un heterodímero de las subunidades catalíticas de IKK alfa e IKK beta y por una proteína reguladora “maestra” llamada NEMO (modulador esencial NF-κB) o IKK gamma. Cuando es activada por señales, normalmente procedentes de fuera de la célula, la IκB quinasa fosforila dos residuos de serina localizados en un dominio IκB regulador. Una vez fosforiladas estas serinas (por ejemplo, las serinas 32 y 36 en la IκBα humana), las moléculas del inhibidor IκB son modificadas por un proceso llamado ubiquitinación, que después las lleva a ser degradadas por una estructura celular llamada proteosoma.
Con la degradación de IκB, el complejo NF-κB es libre para entrar al núcleo dónde puede activar la expresión de los genes específicos que tienen cerca sitios de unión de ADN para NFκB. La activación de estos genes por NF-κB entonces induce una respuesta fisiológica, como por ejemplo, una respuesta inflamatoria o inmune, una respuesta de supervivencia celular, o una proliferación celular. NFκB activa la expresión de su propio represor, IκBα. El nuevamente sintetizado IκBα re-inhibe NFκB y, por tanto, forma un bucle de auto feedback, que provoca niveles oscilatorios de la actividad de NFκB.[31] Además, varios virus, incluyendo el virus VIH del SIDA, poseen sitios de unión para NFκB que controlan la expresión de genes virales, que a su vez contribuyen a la replicación viral o a la patogenicidad viral. En el caso de VIH-1, la activación de NFκB puede verse involucrada, al menos en parte, en la activación del virus desde un estado latente e inactivo.[32] YopP es un factor secretado por la Yersinia pestis, el agente causante de la peste, que previene la ubiquitinación de IκB. Esto provoca que este agente patógeno inhiba eficazmente la vía NF- κB y así bloquea la respuesta inmune de una persona infectada con Yersinia.[33]
Inhibidores de la actividad NF-κB
En cuanto a las proteínas inhibidoras de la actividad NF-κB, una de ellas, IFRD1, reprime la actividad de la p65 de NF-κB mediante la mejora de la desacetilación mediada por HDAC de la subunidad p65 a lisina 310, favoreciendo el secuestro de la HDAC3 a p65. De hecho, IFRD1 forma complejos trimoleculares con p65 y HDAC3.[34][35]
No canónico
Un selecto grupo de estímulos de diferenciación celular o desarrollo celular, tales como linfotoxina-α, BAFF o RANKL, activan la vía no canónica de NF-κB para inducir el dímero NF-κB/RelB:p52 en el núcleo. En esta vía, la activación de la quinasa inductora de NF-κB (NIK) sobre el ligando del receptor induce la fosforilación y el posterior procesamiento proteosomal de la proteína precursora de NF-κB2 de p100 a subunidad p52 madura a través de una vía IKK1/IKKa dependiente. Entonces la p52 dimeriza con RelB para aparecer como RelB:p52 nuclear con actividad de unión al ADN para regular una clase distinta de genes.[36] En contraste con la señalización canónica, que responde a la degradación mediada por NEMO-IKK2 de IκBα, -β, -ε, la señalización no canónica difícilmente depende del proceso de p100 a p52 mediado por NIK. Teniendo en cuenta sus diferentes regulaciones, se pensaba que estas dos vías eran independientes una de otra. No obstante, recientes análisis han revelado que la síntesis de los constituyentes de la vía no canónica, es decir RelB y p52, es controlada por la señalización canónica de IKK2-IκB-RelA:p50.[37] Por otra parte, la generación de los dímeros tanto canónicos como no canónicos, es decir RelA:p50 y RelB:p52, en el medio celular, también están relacionadas entre sí de manera mecánica.[37] Estos análisis sugieren que un sistema NF-κB integrado conduce a la activación de los dímeros RelA y RelB y que un mal funcionamiento de la vía canónica puede dar lugar a una respuesta celular aberrante también a través de la vía no canónica.
Inmunidad
El NF-κB2 es un factor de transcripción principal que regula genes responsables tanto de la respuesta innata como de la respuesta adaptativa. Tras la activación de los receptores de los linfocitos T o B, el factor NF-κB se activa a través de distintos componentes de señalización. El receptor de la célula T, la proteína quinasa Lck, es reclutada y fosforilada por los motivos ITAM que se encuentran en el citoplasma de CD3. A continuación la molécula ZAP es reclutada y fosforilada por ITAM que ayudan a recluir LAT y PLC gamma, las cuales causan la activación de PKC: a través de una cascada de eventos de fosforilación, el complejo quinasa se activa y NF-κB es capaz de entrar en el núcleo para regular al azar los genes implicados en el desarrollo, maduración y proliferación de las células T.[38]
En neuronas
Además de las funciones en la supervivencia celular, el NF-κB tiene diversas funciones en el sistema nervioso, incluyendo la plasticidad, el aprendizaje y la memoria neuronal. Además de los estímulos que activan el NF-κB en otros tejidos, en el sistema nervioso puede ser activado por factores de Crecimiento (BDNF, NGF) utilizando el glutamato como neurotransmisor.[8] Estos activadores de NF-κB en el sistema nervioso convergen en el complejo de IKK y la vía canónica.
Recientemente se ha mostrado un gran interés por el papel de NF-κB en el sistema nervioso. Los estudios actuales sugieren que el NF-κB es importante para el aprendizaje y la memoria en múltiples organismos como cangrejos,[10][11] moscas de la fruta,[39] y ratones.[8][9] El factor NF-κB puede regular el aprendizaje y la memoria, en parte, por la modulación de la plasticidad,[7][40] función sináptica,[39][41][42] así como el crecimiento de las dendritas[43] y de las espinas dendríticas.[42]
Se ha demostrado que los genes que disponen de sitios de unión para el NF-κB, tienen un mayor aprendizaje en la siguiente transcripción genética,[8] lo que sugiere que este factor en el sistema nervioso es importante para la plasticidad y el aprendizaje. Muchos de estos genes diana de NF-κB son los receptores de glutamato (AMPA y NMDA-R-R),[44][45][46][47] los factores de crecimiento (BDNF, NGF),[48] citoquinas (TNF-alfa, TNFR),[49] quinasas (PKAc),[40] y las proteínas de unión sináptica (PSD-95).[42]
Importancia clínica
Descripción general de las vías de transducción de señales implicadas en la apoptosis. El NF-κB es ampliamente utilizado por las células eucariotas como regulador de los genes que controlan la proliferación celular y la supervivencia celular. Debido a ello, muchos tipos diferentes de tumores humanos tienen mal regulados el NF-κB (es decir, activado). Cuando NF-κB se encuentra activado, induce la expresión de los genes que protegen e inducen la proliferación de células que en otras condiciones deberían morir por apoptosis para evitar que el daño se extienda.
Los defectos en NF-κB producen una mayor susceptibilidad a la apoptosis que conduce que haya un aumento en la muerte celular . Esto se debe a que NF-κB regula genes anti-apoptóticos (sobre todo la TRAF1 TRAF2) y por tanto, controla la actividad enzimática de las caspasas, que son fundamentales para la mayoría de los procesos apoptóticos.[50]
En las células tumorales, NF-κB se activa, ya sea debido a mutaciones en genes que codifican los factores de transcripción de NF-κB o en los genes que controlan la actividad de NF-κB (por ejemplo, genes IkB) y, además, algunas células tumorales secretan factores que son activadores de NF -kB. El bloqueo de NF-κB puede causar que las células tumorales dejen de proliferar, morir, o que sean más sensibles a la acción de los agentes anti-tumorales. Por lo tanto, NF-κB es el objeto de muchas empresas farmacéuticas para la terapia contra el cáncer.[51]
Debido a que NF-κB controla varios genes involucrados en la inflamación, no es de extrañar que NF-κB se encuentre activado crónicamente en enfermedades inflamatorias, tales como la enfermedad inflamatoria intestinal, la artritis, sepsis, gastritis, asma y arterosclerosis entre otros.[52] Es importante señalar que los reguladores clave de la NF-κB se asocian con una elevada mortalidad, especialmente en enfermedades cardiovasculares[53][54] y esquizofrénicas.[55]
Muchos productos naturales , incluidos los anti-oxidantes que tienen actividad anticancerígena y antiinflamatoria , se ha demostrado que también son capaces de inhibir NF-κB. Hay una polémica patente en los EE.UU[56] que aplica el descubrimiento y el uso de agentes que pueden bloquear la NF-κB con fines terapéuticos. Esta patente está involucrada en varios juicios, incluyendo Ariad v Lilly. Un trabajo reciente de Karin,[57] Ben-Neriah[58] y otros han remarcado la importancia de la conexión entre el NF-κB, la inflamación y el cáncer, dando una mayor importancia a las terapias inhibitorias de NF-κB.[59]
Extractos procedentes de un gran número de plantas también son inhibidores de la activación de NF-κB in vitro.[60]
Área terapéutica
Se ha observado que en muchos cánceres hay una sobre activación del factor NF-κB. Por otra parte, la supresión de NF-κB limita la proliferación de células cancerosas. Además, NF-κB juega un papel clave en la respuesta inflamatoria. Por lo tanto, los métodos de inhibición de señalización de NF-κB tienen gran interés terapéutico en cánceres y enfermedades inflamatorias.[61][62]
La activación de la translocación nuclear de NF-κB es independiente al aumento de estrés oxidativo,[63] por lo que, gracias a ello, se puede llevar a cabo un mejor estudio de posibles estrategias mediante inhibición de NF-κB.
Un nuevo medicamento llamado denosumab actúa aumentando la densidad mineral ósea y, por lo tanto, reduciendo las fracturas óseas en muchos pacientes. Inhibe la RANKL, que actúa a través de su receptor RANK, que a su vez promueve la NF-κB,[64] RANKL normalmente permite la diferenciación de los osteoclastos a partir de monocitos.
Otros medicamentos son el disulfiram, olmesartán y ditiocarbamatos, que inhiben la cascada de señalización del factor nuclear κB (NF-κB).[65]
Carcinoma Nasofaríngeo, EBV y NFKB
El Carcinoma Nasofaríngeo (NPC) se caracteriza por células epiteliales tumorales poco diferenciadas que residen en la parte posterior a la faringe. En este cuadro clínico se produce un microambiente complejo con gran infiltración de linfocitos, dando lugar a la aparición de un linfoepitelioma con un aparente fenotipo inflamatorio. Además en un 90% de los casos las células malignas son uniformemente positivas para el virus de Epstein-Barr (VEB), un herpes virus muy extendido en la población humana.
Estudios funcionales y genómicos recientes han implicado la activación de la vía NF-κB y la evasión inmunitaria en este tipo de carcinoma nasofaringeo asociados a EBV. En estos se muestran una activación constitutiva de las vías inflamatorias de NF-κB en un 90% de los casos que padecen Carcinoma Nasofaríngeo (NPC) asociados al virus de Epstein-Barr, ya sea a través de alteraciones somáticas o a través de la expresión del oncogén LMP1 viral codificado por el virus. Esta señalización aberrante de NF-κB ayuda a la progresión de este tipo de cáncer asociado a EBV, decrito como una característica genómica.[66] En cuanto a la evasión de sistema inmune, se ha decrito que el 91,4% de los casos de carcinoma Nasofaringeo positivo para EBV, padece de alteraciones somáticas o sobreexpresión de varios genes virales (LMP1, BNLF2a) dirigidos a la inmunidad innata y adaptativa. Es probable que esta característica surja para contrarrestar el entorno inflamatorio debido a la infección persistente por el virus.[66]
Véase también
- IKK2
Referencias
- Gilmore TD (2006). «Introduction to NF-κB: players, pathways, perspectives». Oncogene 25 (51): 6680-4. PMID 17072321. doi:10.1038/sj.onc.1209954.
- Brasier AR (2006). «The NF-κB regulatory network». Cardiovasc. Toxicol. 6 (2): 111-30. PMID 17303919. doi:10.1385/CT:6:2:111.
- Perkins ND (enero de 2007). «Integrating cell-signalling pathways with NF-κB and IKK function». Nat. Rev. Mol. Cell Biol. 8 (1): 49-62. PMID 17183360. doi:10.1038/nrm2083.
- Concetti, Julia; Wilson, Caroline L. (7 de septiembre de 2018). «NFKB1 and Cancer: Friend or Foe?». Cells 7 (9). ISSN 2073-4409. PMC 6162711. PMID 30205516. doi:10.3390/cells7090133.
- Gilmore TD (1999). «The Rel/NF-κB signal transduction pathway: introduction». Oncogene 18 (49): 6842-4. PMID 10602459. doi:10.1038/sj.onc.1203237.
- Tian B, Brasier AR (2003). «Identification of a nuclear factor κ B-dependent gene network». Recent Prog. Horm. Res. 58: 95-130. PMID 12795416. doi:10.1210/rp.58.1.95.
- Albensi BC, Mattson MP (2000). «Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity». Synapse 35 (2): 151-9. PMID 10611641. doi:10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P.
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (octubre de 2003). «NF-kappa B functions in synaptic signaling and behavior». Nat. Neurosci. 6 (10): 1072-8. PMID 12947408. doi:10.1038/nn1110.
- Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, Pizzi M, Liou HC, Sweatt JD (abril de 2004). «A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel». J. Neurosci. 24 (16): 3933-43. PMID 15102909. doi:10.1523/JNEUROSCI.5646-03.2004.
- Freudenthal R, Locatelli F, Hermitte G, Maldonado H, Lafourcade C, Delorenzi A, Romano A (febrero de 1998). «Kappa-B like DNA-binding activity is enhanced after spaced training that induces long-term memory in the crab Chasmagnathus». Neurosci. Lett. 242 (3): 143-6. PMID 9530926. doi:10.1016/S0304-3940(98)00059-7.
- Merlo E, Freudenthal R, Romano A (2002). «The IkappaB kinase inhibitor sulfasalazine impairs long-term memory in the crab Chasmagnathus». Neuroscience 112 (1): 161-72. PMID 12044481. doi:10.1016/S0306-4522(02)00049-0.
- Sen R, Baltimore D (1986). «Multiple nuclear factors interact with the immunoglobulin enhancer sequences». Cell 46 (5): 705-16. PMID 3091258. doi:10.1016/0092-8674(86)90346-6.
- Karin M, Ben-Neriah Y (2000). «Phosphorylation meets ubiquitination: the control of NF-κB activity». Annu. Rev. Immunol. 18: 621-63. PMID 10837071. doi:10.1146/annurev.immunol.18.1.621.
- Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M (2001). «Activation by IKKalpha of a second, evolutionary conserved, NF-κB signaling pathway». Science 293 (5534): 1495-9. PMID 11520989. doi:10.1126/science.1062677.
- Nabel GJ, Verma IM (noviembre de 1993). «Proposed NF-κB/IκB family nomenclature». Genes Dev. 7 (11): 2063. PMID 8224837. doi:10.1101/gad.7.11.2063.
- Ghosh S, May MJ, Kopp EB (1998). «NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses». Annu. Rev. Immunol. 16: 225-60. PMID 9597130. doi:10.1146/annurev.immunol.16.1.225.
- Waterhouse RM, Kriventseva EV, Meister S, Xi Z, Alvarez KS, Bartholomay LC, Barillas-Mury C, Bian G, Blandin S, Christensen BM, Dong Y, Jiang H, Kanost MR, Koutsos AC, Levashina EA, Li J, Ligoxygakis P, Maccallum RM, Mayhew GF, Mendes A, Michel K, Osta MA, Paskewitz S, Shin SW, Vlachou D, Wang L, Wei W, Zheng L, Zou Z, Severson DW, Raikhel AS, Kafatos FC, Dimopoulos G, Zdobnov EM, Christophides GK (2007). «Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes». Science 316 (5832): 1738-43. PMC 2042107. PMID 17588928. doi:10.1126/science.1139862.
- PDB 3do7 ; Fusco AJ, Huang DB, Miller D, Wang VY, Vu D, Ghosh G (febrero de 2009). «NF-kappaB p52:RelB heterodimer recognizes two classes of kappaB sites with two distinct modes». EMBO Rep. 10 (2): 152-9. PMC 2637311. PMID 19098713. doi:10.1038/embor.2008.227.
- (a) Chandel NS, Trzyna WC, McClintock DS, Schumacker PT (julio de 2000). «Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin». J Immunol 165 (2): 1013-1021. PMID 10878378.; (b) Fitzgerald DC, Meade KG, McEvoy AN, Lillis L, Murphy EP, MacHugh DE, Baird AW (marzo de 2007). «Tumour necrosis factor-alpha (TNF-alpha) increases nuclear factor kappaB (NFkappaB) activity in and interleukin-8 (IL-8) release from bovine mammary epithelial cells». Vet Immunol Immunopathol 116 (1-2): 59-68. PMID 17276517. doi:10.1016/j.vetimm.2006.12.008.; (c) Renard P, Zachary MD, Bougelet C, Mirault ME, Haegeman G, Remacle J, Raes M (enero de 1997). «Effects of antioxidant enzyme modulations on interleukin-1-induced nuclear factor kappa B activation». Biochem Pharmacol 53 (2): 149-160. PMID 9037247.; (d) Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN (noviembre de 2005). «LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia». Blood 106 (9): 3114-3122. PMC 1895321. PMID 16020513. doi:10.1182/blood-2005-02-0759.; (e) Takemoto Y, Yoshiyama M, Takeuchi K, Omura T, Komatsu R, Izumi Y, Kim S, Yoshikawa J (noviembre de 1999). «Increased JNK, AP-1 and NF-kappa B DNA binding activities in isoproterenol-induced cardiac remodeling». J Mol Cell Cardiol 31 (11): 2017-2030. PMID 10591028. doi:10.1006/jmcc.1999.1033.; (f) Hargrave BY, Tiangco DA, Lattanzio FA, Beebe SJ (2003). «Cocaine, not morphine, causes the generation of reactive oxygen species and activation of NF-kappaB in transiently cotransfected heart cells». Cardiovasc Toxicol 3 (2): 141-151. PMID 14501032. doi:10.1385/CT:3:2:141.; (g) Basu S, Rosenzweig KR, Youmell M, Price BD (junio de 1998). «The DNA-dependent protein kinase participates in the activation of NF kappa B following DNA damage». Biochem Biophys Res Commun 247 (1): 79-83. PMID 9636658. doi:10.1006/bbrc.1998.8741.
- Baud'huin M, Lamoureux F, Duplomb L, Rédini F, Heymann D (septiembre de 2007). «RANKL, RANK, osteoprotegerin: key partners of osteoimmunology and vascular diseases». Cell Mol Life Sci 64 (18): 2334-2350. PMID 17530461. doi:10.1007/s00018-007-7104-0.
- Doyle SL, O'Neill LA (octubre de 2006). «Toll-like receptors: from the discovery of NFκB to new insights into transcriptional regulations in innate immunity». Biochem. Pharmacol. 72 (9): 1102-13. PMID 16930560. doi:10.1016/j.bcp.2006.07.010.
- Hayden MS, West AP, Ghosh S (octubre de 2006). «NF-κB and the immune response». Oncogene 25 (51): 6758-80. PMID 17072327. doi:10.1038/sj.onc.1209943.
- Li Q, Verma IM (2002). «NF-κB regulation in the immune system». Nat. Rev. Immunol. 2 (10): 725-34. PMID 12360211. doi:10.1038/nri910.
- Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D (1993). «The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers». Genes Dev. 7 (7B): 1354-63. PMID 8330739. doi:10.1101/gad.7.7b.1354.
- Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U (1992). «The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB-mediated inhibition». Nature 359 (6393): 339-42. PMID 1406939. doi:10.1038/359339a0.
- Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U (1993). «The oncoprotein Bcl-3 directly transactivates through κ B motifs via association with DNA-binding p50B homodimers». Cell 72 (5): 729-39. PMID 8453667. doi:10.1016/0092-8674(93)90401-B.
- Jacobs MD, Harrison SC (1998). «Structure of an IκBalpha/NF-κB complex». Cell 95 (6): 749-58. PMID 9865693. doi:10.1016/S0092-8674(00)81698-0.
- Basak S, Kim H, Kearns JD, Tergaonkar V, O'dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A (2007). «A fourth IκB protein within the NF-κB signaling module». Cell 128 (2): 369-81. PMC 1831796. PMID 17254973. doi:10.1016/j.cell.2006.12.033..
- Dobrzanski P, Ryseck RP, Bravo R (1995). «Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100». Oncogene 10 (5): 1003-7. PMID 7898917.
- Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, Weih F, Franzoso G, Hoffmann A, Fu YX (2006). «Coordination between NF-κB family members p50 and p52 is essential for mediating LTbetaR signals in the development and organization of secondary lymphoid tissues». Blood 107 (3): 1048-55. PMC 1895903. PMID 16195333. doi:10.1182/blood-2005-06-2452.
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR (2004). «Oscillations in NF-κB signaling control the dynamics of gene expression». Science 306 (5696): 704-8. PMID 15499023. doi:10.1126/science.1099962.
- Hiscott J, Kwon H, Génin P (enero de 2001). «Hostile takeovers: viral appropriation of the NF-κB pathway». J. Clin. Invest. 107 (2): 143-51. PMC 199181. PMID 11160127. doi:10.1172/JCI11918.
- Adkins I, Schulz S, Borgmann S, Autenrieth IB, Gröbner S (febrero de 2008). «Differential roles of Yersinia outer protein P-mediated inhibition of nuclear factor-κB in the induction of cell death in dendritic cells and macrophages». J. Med. Microbiol. 57 (Pt 2): 139-44. PMID 18201977. doi:10.1099/jmm.0.47437-0.
- Micheli L, Leonardi L, Conti F, Buanne P, Canu N, Caruso M, Tirone F (marzo de 2005). «PC4 coactivates MyoD by relieving the histone deacetylase 4-mediated inhibition of myocyte enhancer factor 2C». Mol. Cell. Biol. 25 (6): 2242-59. PMC 1061592. PMID 15743821. doi:10.1128/MCB.25.6.2242-2259.2005.
- Micheli L, Leonardi L, Conti F, Maresca G, Colazingari S, Mattei E, Lira SA, Farioli-Vecchioli S, Caruso M, Tirone F (febrero de 2011). «PC4/Tis7/IFRD1 stimulates skeletal muscle regeneration and is involved in myoblast differentiation as a regulator of MyoD and NF-kappaB». J. Biol. Chem. 286 (7): 5691-707. PMC 3037682. PMID 21127072. doi:10.1074/jbc.M110.162842.
- Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M (2004). «Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers». EMBO J 23 (21): 4202-10. PMID 11547050. doi:10.1038/sj.emboj.7600391.
- Basak S, Shih VF, Hoffmann A (2008). «Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system». Mol Cell Biol 28 (10): 3139-50. PMC 2423155. PMID 18299388. doi:10.1128/MCB.01469-07.
- Livolsi A, Busuttil V, Imbert V, Abraham RT, Peyron JF (marzo de 2001). «Tyrosine phosphorylation-dependent activation of NF-κB. Requirement for p56 LCK and ZAP-70 protein tyrosine kinases». Eur. J. Biochem. 268 (5): 1508-15. PMID 11231305. doi:10.1046/j.1432-1327.2001.02028.x.
- Heckscher ES, Fetter RD, Marek KW, Albin SD, Davis GW (septiembre de 2007). «NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ». Neuron 55 (6): 859-73. PMC 2701504. PMID 17880891. doi:10.1016/j.neuron.2007.08.005.
- Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prüllage M, Pfeiffer J, Lindecke A, Staiger V, Israël A, Kaltschmidt C, Mémet S (abril de 2006). «NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling». Mol. Cell. Biol. 26 (8): 2936-46. PMC 1446931. PMID 16581769. doi:10.1128/MCB.26.8.2936-2946.2006.
- Wang J, Fu XQ, Lei WL, Wang T, Sheng AL, Luo ZG (agosto de 2010). «Nuclear factor kappaB controls acetylcholine receptor clustering at the neuromuscular junction». J. Neurosci. 30 (33): 11104-13. PMID 20720118. doi:10.1523/JNEUROSCI.2118-10.2010.
- Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK (April de 2011). «A Requirement for Nuclear Factor-{kappa}B in Developmental and Plasticity-Associated Synaptogenesis». J. Neurosci. 31 (14): 5414-25. PMID 21471377. doi:10.1523/JNEUROSCI.2456-10.2011.
- Gutierrez H, Hale VA, Dolcet X, Davies A (abril de 2005). «NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS». Development 132 (7): 1713-26. PMID 15743881. doi:10.1242/dev.01702.
- Borges K, Dingledine R (julio de 2001). «Functional organization of the GluR1 glutamate receptor promoter». J. Biol. Chem. 276 (28): 25929-38. PMID 11340067. doi:10.1074/jbc.M009105200.
- Yu Z, Cheng G, Wen X, Wu GD, Lee WT, Pleasure D (octubre de 2002). «Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NF-kappaB-dependent mechanism». Neurobiol. Dis. 11 (1): 199-213. PMID 12460558. doi:10.1006/nbdi.2002.0530.
- Richter M, Suau P, Ponte I (julio de 2002). «Sequence and analysis of the 5' flanking and 5' untranslated regions of the rat N-methyl-D-aspartate receptor 2A gene». Gene 295 (1): 135-42. PMID 12242020. doi:10.1016/S0378-1119(02)00833-8.
- Begni S, Moraschi S, Bignotti S, Fumagalli F, Rillosi L, Perez J, Gennarelli M (abril de 2003). «Association between the G1001C polymorphism in the GRIN1 gene promoter region and schizophrenia». Biol. Psychiatry 53 (7): 617-9. PMID 12679240. doi:10.1016/S0006-3223(02)01783-3.
- Zaheer A, Yorek MA, Lim R (diciembre de 2001). «Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion». Neurochem. Res. 26 (12): 1293-9. PMID 11885780. doi:10.1023/A:1014241300179.
- Qiu J, Hu X, Nesic O, Grafe MR, Rassin DK, Wood TG, Perez-Polo JR (julio de 2004). «Effects of NF-kappaB oligonucleotide "decoys" on gene expression in P7 rat hippocampus after hypoxia/ischemia». J. Neurosci. Res. 77 (1): 108-18. PMID 15197744. doi:10.1002/jnr.20156.
- Sheikh MS, Huang Y (2003). «Death receptor activation complexes: it takes two to activate TNF receptor 1». Cell Cycle 2 (6): 550-2. PMID 14504472.
- Escárcega RO, Fuentes-Alexandro S, García-Carrasco M, Gatica A, Zamora A (2007). «The transcription factor nuclear factor-κB and cancer». Clinical Oncology (Royal College of Radiologists (Great Britain)) 19 (2): 154-61. PMID 17355113. doi:10.1016/j.clon.2006.11.013.
- Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, Cheshire N, Paleolog E, Feldmann M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A. 2004 Apr 13;101(15):5634-9. Epub 2004 Apr 2. PubMed PMID 15064395 Link to paper;; PubMed Central PMCID: PMC397455.
- Venuraju SM, Yerramasu A, Corder R, Lahiri A (mayo de 2010). «Osteoprotegerin as a predictor of coronary artery disease and cardiovascular mortality and morbidity». J. Am. Coll. Cardiol. 55 (19): 2049-61. PMID 20447527. doi:10.1016/j.jacc.2010.03.013.
- Lieb W, Gona P, Larson MG, Massaro JM, Lipinska I, Keaney JF, Rong J, Corey D, Hoffmann U, Fox CS, Vasan RS, Benjamin EJ, O'Donnell CJ, Kathiresan S (septiembre de 2010). «Biomarkers of the osteoprotegerin pathway: clinical correlates, subclinical disease, incident cardiovascular disease, and mortality». Arterioscler. Thromb. Vasc. Biol. 30 (9): 1849-54. PMC 3039214. PMID 20448212. doi:10.1161/ATVBAHA.109.199661.
- Song XQ, Lv LX, Li WQ, Hao YH, Zhao JP (marzo de 2009). «The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia». Biol. Psychiatry 65 (6): 481-8. PMID 19058794. doi:10.1016/j.biopsych.2008.10.018.
- Plantilla:US patent reference
- Karin M (marzo de 2008). «The IκB kinase - a bridge between inflammation and cancer». Cell Res. 18 (3): 334-42. PMID 18301380. doi:10.1038/cr.2008.30.
- Pikarsky E, Ben-Neriah Y (abril de 2006). «NF-κB inhibition: a double-edged sword in cancer?». Eur. J. Cancer 42 (6): 779-84. PMID 16530406. doi:10.1016/j.ejca.2006.01.011.
- Mantovani A, Marchesi F, Portal C, Allavena P, Sica A (2008). «Linking inflammation reactions to cancer: novel targets for therapeutic strategies». Adv. Exp. Med. Biol. 610: 112-27. PMID 18593019. doi:10.1007/978-0-387-73898-7_9.
- Paur I, Balstad TR, Kolberg M, Pedersen MK, Austenaa LM, Jacobs DR, Blomhoff R (mayo de 2010). «Extract of oregano, coffee, thyme, clove, and walnuts inhibits NF-kappaB in monocytes and in transgenic reporter mice». Cancer Prev Res (Phila) 3 (5): 653-63. PMID 20424131. doi:10.1158/1940-6207.CAPR-09-0089.
- Garg A, Aggarwal BB (junio de 2002). «Nuclear transcription factor-kappaB as a target for cancer drug development». Leukemia 16 (6): 1053-68. PMID 12040437. doi:10.1038/sj.leu.2402482.
- Sethi G, Sung B, Aggarwal BB (enero de 2008). «Nuclear factor-kappaB activation: from bench to bedside». Exp. Biol. Med. (Maywood) 233 (1): 21-31. PMID 18156302. doi:10.3181/0707-MR-196.
- Vlahopoulos S, Boldogh I, Casola A, Brasier AR (septiembre de 1999). «Nuclear factor-κB-dependent induction of interleukin-8 gene expression by tumor necrosis factor alpha: evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation». Blood 94 (6): 1878-89. PMID 10477716. Archivado desde el original el 12 de septiembre de 2019. Consultado el 18 de noviembre de 2011.
- Hamdy NA (2008). «Denosumab: RANKL inhibition in the management of bone loss». Drugs Today (Barc). 44 (1): 7-21. PMID 18301800. doi:10.1358/dot.2008.44.1.1178467.
- Cvek B, Dvorak Z (2007). «Targeting of nuclear factor-κB and proteasome by dithiocarbamate complexes with metals». Curr. Pharm. Des. 13 (30): 3155-67. PMID 17979756. doi:10.2174/138161207782110390.
- {{Bruce JP, To KF, Lui VWY, et al. El perfil del genoma completo del carcinoma nasofaríngeo revela la cooperación virus-huésped en la activación inflamatoria de NF-κB y el escape inmune. Nat Commun. 2021;12(1):4193. Publicado 2021 Jul 7. doi:10.1038/s41467-021-24348-6}}
Enlaces externos
- MeSH: NF-kappa+B (en inglés)
- Sankar Ghosh (2006). Handbook of Transcription Factor NF-κB. Boca Raton: CRC. ISBN 0-8493-2794-6.
- Thomas D Gilmore. «The Rel/NF-κB Signal Transduction Pathway». Boston University. Consultado el 2 de diciembre de 2007.
- Traducción de en:NF-κB (versión: http://en.wikipedia.org/wiki/NF-%CE%BAB)
| Control de autoridades |
|
|---|
 Datos: Q411114
Datos: Q411114 Multimedia: NF-kappa B / Q411114
Multimedia: NF-kappa B / Q411114