Galactosemia
La galactosemia es una enfermedad hereditaria causada por una deficiencia enzimática y se manifiesta con incapacidad de utilizar el azúcar simple galactosa, lo cual provoca una acumulación de este dentro del organismo, produciendo lesiones en el hígado y el sistema nervioso central.
| Galactosemia | ||
|---|---|---|
| Especialidad | endocrinología | |

Concepto
La galactosemia es una enfermedad caracterizada por la incapacidad de metabolizar la galactosa en glucosa. Esto se debe a que el sujeto hereda un gen defectuoso de cada progenitor. La galactosa es un monosacárido obtenido principalmente de la hidrólisis de la lactosa contenida en la leche, aunque también puede estar presente en otros alimentos. La galactosa se absorbe en el intestino y principalmente se transforma en glucosa en el hígado.
Existe una deficiencia en la enzima galactosa-1-fosfato uridiltransferasa, que es imprescindible para pasar de galactosa a glucosa. Normalmente cuando una persona consume un producto que contiene lactosa, el metabolismo degrada la lactosa en galactosa y luego a glucosa. Una cantidad excesiva de galactosa en sangre causa la dicha galactosemia. Esta se caracteriza por causar daños en el hígado, riñones y sistema nervioso central, entre otros sistemas.
Existe una forma de menor gravedad de galactosemia, que se debe a la deficiencia de galactoquinasa. Esta variante, por el contrario, se puede tratar a base de una dieta estricta y no provoca daños hepáticos y/o neurológicos.
Tal y como se verá a continuación, hay varios tipos de galactosemia, causadas por el déficit de distintas enzimas. Las consecuencias de esta enfermedad pueden llegar a ser mortales, por lo que es muy importante una detección precoz del problema. Además, requiere un buen tratamiento para superar esta enfermedad.
Genética
La galactosemia sigue un patrón de herencia mendeliana de tipo autosómico recesivo, eso quiere decir que para que el sujeto en cuestión presente la enfermedad, ha de presentar dos copias de un gen anormal. Además, la penetrancia en homocigosis es completa.
Si ambos padres son portadores sanos heterocigotos, estos transmitirán la enfermedad a un 25% de sus hijos, que tendrán dos copias del gen de la enfermedad y habrán heredado una copia del gen alterado de cada uno de sus progenitores. Otro 25% de los hijos serán totalmente sanos (no padecerán ni serán portadores de la enfermedad); y finalmente un 50% de los descendientes serán portadores asintomáticos. Así pues, una persona con un solo alelo, se dice que es “portador” pero no padece la enfermedad, sin embargo, este puede pasar el gen anormal a sus descendientes, y estos sí manifestarlo.
Los dos tipos de galactosemia más tratados son: la galactosemia clásica y la galactosemia Duarte. La galactosemia clásica se debe a que el individuo no sintetiza la enzima GALT por lo que no puede degradar la galactosa. En la galactosemia Duarte, por el contrario, sí se sintetiza la enzima GALT, pero de forma deficiente (véase más adelante).
En resumen, generalmente los padres portadores asintomáticos tendrán un 25% de los hijos sanos, un 50% de los hijos portadores asintomáticos y un 25% de los hijos con galactosemia.
Sin embargo, pueden darse otras combinaciones genéticas posibles entre parentales, que cambiaría la distribución en la descendencia. Por ejemplo:
- Padre enfermo × Madre portadora: el 50% de los hijos serán homocigóticos para la mutación (es decir, enfermos), y el otro 50% será portador.
- Padre enfermo × Madre sana: todos sus descendientes serán portadores, pero ninguno de ellos presentará la enfermedad.
- Padre portador × Madre sana: el 50% de los hijos serán portadores pero ninguno de ellos manifestará la enfermedad.
Cabe mencionar que hay muy pocos casos de mujeres enfermas que queden embarazadas, debido a la alta frecuencia a la que estas sufren atrofia ovárica e hipogonadismo.
Bioquímica
Durante la digestión de la lactosa, la enzima lactasa degrada la molécula en glucosa y galactosa. Los individuos que padecen galactosemia, tienen niveles muy bajos o ausencia completa de las enzimas necesarias para la posterior metabolización de la galactosa, lo que conlleva la acumulación de galactosa 1- fosfato en diversos tejidos. Esta acumulación genera niveles tóxicos de galactosa que, tal y como sucede en el tipo clásico de este trastorno, pueden provocar hepatomegalia (un aumento patológico del hígado), cirrosis, fallo renal, cataratas, daños cerebrales y, en mujeres, disfunción ovárica. El tratamiento de esta enfermedad es crucial puesto que la mortalidad en niños con galactosemia sin tratamiento es de un 75%. Más adelante se comenta el tratamiento de esta enfermedad.

Metabolismo de la galactosa
La metabolización de la galactosa a glucosa se realiza a través de la vía Leloir, la cual implica una serie de reacciones enzimáticas realizadas por diferentes enzimas. Si la galactosa no es metabolizada por la vía Leloir, explicada a continuación, puede metabolizarse por dos vías alternativas: En la primera la galactosa es reducida por una aldosa reductasa a galactitol (Reacción 6), mientras que en la segunda la galactosa es oxidada por una galactosa deshidrogenasa a galactonato (Reacción 7).

Vía de Leloir
La transformación de galactosa en glucosa se da a través de una serie de reacciones que conforman la vía de Leloir[1] (Reacciones 1, 2, 8 y 10), para la que son necesarias tres enzimas: la galactoquinasa (GALK), la galactosa-1-P uridiltransferasa (GALT) y la UDP-galactosa 4-epimerasa (GALE).[2] El papel biológico de estas enzimas es permitir la interconversión de grupos galactosil y glucosil.
Para convertir la galactosa en glucosa, previamente la enzima galactosa mutorotasa facilita la conversión de la β-D-galactosa en α-D-galactosa, la forma activa para la vía.

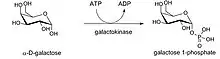
A continuación, tiene lugar la fosforilación de la galactosa mediante la enzima GALK, produciendo así galactosa-1-fosfato. Esta reacción consume una molécula de ATP.
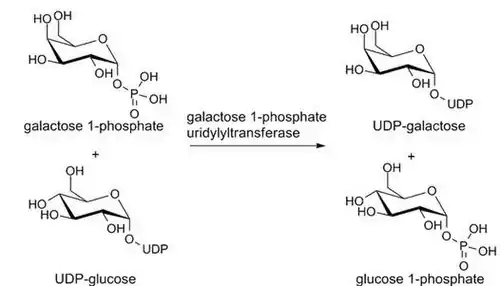
En el siguiente paso, la enzima GALT convierte la galactosa 1-fosfato en UDP-galactosa usando UDP-glucosa como fuente de uridina difosfato (UDP).
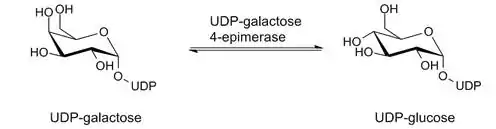
Finalmente, la enzima GALE recicla la UDP-galactosa a UDP-glucosa para la posterior reacción de transferasa, donde se transferirá el grupo funcional y de la cual se liberará glucosa-1-fosfato.
Adicionalmente, la glucosa-1-fosfato es transformada en glucosa-6-fosfato y así puede entrar en la glicólisis para ser metabolizada y generar energía.
Degradación a galactitol
En pacientes con galactosemia, la acumulación de galactosa se convierte en el substrato de enzimas que catalizan la vía poliol del metabolismo de los carbohidratos. La primera reacción de esta vía es la reducción de aldosas a polialcoholes. Estudios recientes sugieren que la aldosa-reductasa es la enzima responsable del primer estadio de esta vía. Así pues, la aldosa-reductasa reduce la galactosa a su forma alcohólica, el galactitol. Sin embargo, el galactitol no es un substrato adecuado para la siguiente enzima, polio- deshidrogenasa, lo que provoca o bien la acumulación de galactitol en los tejidos del cuerpo (hígado y cerebro) o bien su excreción a través de la orina.
Oxidación a galactonato
Estudios recientes sugieren que la galactosa-deshidrogenasa es responsable de la conversión de la galactosa en galactonolactona, que después, espontánea o enzimáticamente, se convierte en galactonato. Así pues, la oxidación a galactonato se convierte en una alternativa de la metabolización de la galactosa, siendo la acumulación de galactonato menos perjudicial que la de galactitol.
Tipos
La metabolización de la galactosa para dar glucosa se da gracias a tres enzimas que constituyen lo que se conoce como la vía de Leloir (comentado anteriormente). Los tipos de galactosemia están asociados a las deficiencias de cada uno de los tres tipos de enzimas.
- Deficiencia de galactoquinasa (GALK): en este tipo de galactosemia la galactosa no puede ser fosforilada a galactosa 1-fosfato, por lo que se acumula en los tejidos y se metaboliza por las vías alternativas citadas anteriormente. Los genes mutados que codifican el enzima GALK se encuentran en los cromosomas 15 y 17. Su frecuencia estimada es de 1/150.000-1.000.000 de nacimientos.[3]
- Deficiencia de UDP-galactosa 4-epimerasa (GALE): la reacción que transforma la UDP-galactosa en UDP-glucosa y viceversa no se realiza. El gen mutado que codifica el enzima GALE se encuentra en el cromosoma 1.[4] El gen mutado que codifica el enzima GALE se encuentra en el cromosoma 1.[5]
- Deficiencia de galactosa 1-fosfato uridiltransferasa (GALT): es el tipo más común y grave. También se conoce con el nombre de Galactosemia clásica. En este tipo de galactosemia la galactosa 1-fosfato no puede ser convertida a glucosa 1-fosfato. Se produce acumulación en los tejidos de galactosa y galactosa 1-fosfato. El gen que codifica el enzima GALT se encuentra en el cromosoma 9. Su frecuencia estimada es de 1/40.000-60.000 nacimientos.[6]
| TIPO | GEN AFECTADO | LOCALIZACIÓN CROMOSÓMICA | ENZIMA AFECTADA | NOMBRE DEL TRASTORNO |
|---|---|---|---|---|
| Tipo I | GALT | Cromosoma 9 | Galactosa 1-fosfato uridiltransferasa | Galactosemia clásica |
| Tipo II | GALK1 | Cromosomas 15 y 17 | Galactoquinasa | Déficit de galactoquinasa |
| Tipo III | GALE | Cromosoma 1 | UDP-galactosa 4-epimerasa | Déficit de UDP-galactosa 4-epimerasa |
Síntomas
La sintomatología y su intensidad está determinada por el tipo de deficiencia enzimática que se presente.
Deficiencia de galactoquinasa (GALK). Únicamente se presenta la formación de cataratas debido a la acumulación de galactitol en el cristalino. No hay afectación de hígado, riñones o cerebro. Se caracteriza por un aumento de galactosa y galactitol en plasma y galactosuria.
Deficiencia de UDP-galactosa 4-epimerasa (GALE). Se pueden no mostrar síntomas o presentar síntomas parecidos a los de la galactosemia clásica. En ambos casos se produce una acumulación de UDP-Galactosa y Galactosa 1-fosfato.
Deficiencia de galactosa 1-fosfato uridiltransferasa (GALT). Se presentan letargo, rechazo al alimento y manifestaciones tóxicas generales, incluyendo vómitos y diarreas, pérdida de peso, ictericia, hepatomegalia, ascitis y la formación de cataratas entre otros debido a la acumulación de galactosa, galactitol y galactosa 1-fosfato en los tejidos. También hay un aumento de galactosa y galactitol en plasma, galactosuria e hiperaminoaciduria.
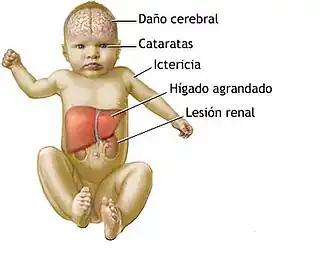
Es importante comenzar cuanto antes el tratamiento ya que pueden aparecer rápidamente nuevos síntomas:
- Convulsiones.
- Insuficiencia hepática y aumento del tamaño del hígado que no funciona adecuadamente.
- Trastornos en la sangre (tales como hemólisis y coagulopatías diversas) que pueden causar choque o la muerte.
- Hipoglucemia y altos niveles de amoníaco que pueden llevar al coma.
Por otra parte, niños con galactosemia leve que necesiten tratamiento y no lo reciban pueden presentar algunos síntomas como:
Diagnóstico
De manera rutinaria los bebés son examinados para ver si presentan galactosemia y es entonces cuando se diagnostica la enfermedad. Los bebés afectados por galactosemia normalmente presentan síntomas de letargo, vómitos, diarrea e ictericia. Ninguno de estos síntomas es específico de la galactosemia, y por eso a menudo hay retrasos en el diagnóstico, especialmente cuando se trata de síntomas leves. Afortunadamente, se diagnostica la mayoría de los casos en la evaluación del recién nacido y si la familia del bebé tiene antecedentes de galactosemia, los médicos pueden comprobarlo antes del nacimiento tomando una muestra del líquido amniótico de alrededor del feto (amniocentesis) o de la placenta (prueba del vello coriónico o CVS).
La prueba de galactosemia es una prueba de sangre (del talón del bebé) o una análisis de orina que busca tres enzimas que son necesarias para cambiar la galactosa en glucosa, azúcar que el cuerpo utiliza para producir energía. Una persona con galactosemia no tiene una de estas enzimas lo cual provoca altos niveles de galactosa en la sangre o en la orina. Es importante que los recién nacidos se sometan a pruebas de trastornos metabólicos sin demora ya que pueden presentar efectos graves, irreversibles o incluso morir en los primeros días de vida.
La galactosemia, también se puede detectar antes de cualquier ingestión de leche materna o fórmula que contenga galactosa. Casi todos los casos de galactosemia clásica pueden detectarse mediante las pruebas de detección sistemática del recién nacido, o NBS. Esta prueba no depende de la ingestión de proteínas o lactosa, y, por lo tanto, se identifica la enfermedad en la primera exploración a menos que el bebé se le haya transferido sangre. Es por eso que la muestra debe tomarse antes de la transfusión. La enzima puede dañarse si el análisis de la muestra se retrasa o es expuesto a altas temperaturas.
Complementariamente, se utilizan dos pruebas para detectar la presencia de la enfermedad:
- Prueba de Beutler: el recién nacido se somete a un cuidado con la inactivación por calor, transfusión o deficiencia de G6PD. La prueba se basa en detectar el nivel de enzimas del bebé, por lo tanto, la ingestión de leche materna u otro producto no afecta el resultado de esta parte de la NBS, y es por eso que la NBS es exacta para detectar galactosemia antes de cualquier ingestión de galactosa.
- Hill test: en esta prueba el bebé no incluye lactosa en su dieta.
De hecho, también se realiza una tercera prueba a todos los que han dado positivo, la llamada “prueba Florida”. El díagnóstico se realiza por:[7]
- Cuantificación de galactosa y galactitol en plasma.
- Cuantificación de galactosa 1-fosfato, galactitol, galactonato y actividad enzimática GALK (galactosa-1-fosfato-uridiltransferasa), GALE y GALT en glóbulos rojos. Si estas enzimas faltan o no hay suficientes, no se puede transformar la galactosa en glucosa y se acumulan grandes cantidades en sangre.
- Deficiencia de la UDP-galactosa-4-epimerasa. Esta causa de galactosemia no es muy habitual y puede resultar sintomática y asintomática. Debe tenerse en cuenta en cualquier paciente con un resultado anormal de galactosa total en el cuerpo y cantidades normales de Gal-1-PUT.
- Presencia de sustancias reductoras en orina como la galactoquinasa, aunque se trata de un raro defecto que sólo se manifiesta si hay desarrollo de cataratas.
- Acumulación de líquido en el abdomen (ascitis).
- Tamaño superior del hígado u otros órganos afectados como el cerebro, los ojos y los riñones.
- Dificultad para crecer o aumentar de peso.
Diagnóstico molecular
Aparte de las pruebas bioquímicas y sintomatológicas, hay otra forma de diagnóstico más sofisticada (aunque también más costosa), que consiste en una prueba molecular. Según los conocimientos actuales, el único gen conocido asociado a la galactosemia es GALT, pero este locus presenta más de 130 posibles mutaciónes causantes de la enfermedad. En la población caucásica predominan dos mutaciones (Q188R y K285N), que están presentes en más del 70% de los casos, mientras que en la población negra existe un principal alelo (S135L), que es responsable del 62% de los casos. Estrictamente, la mayoría de los individuos enfermos son en realidad heterocigotos compuestos en lugar de verdaderos homocigotos, porque poseen dos alelos del gen con diferentes mutaciones. Tanto para la galactosemia clásica como para su variante Duarte, se pueden llevar a cabo tres tipos de pruebas:
- Análisis dirigido de las mutaciones implicadas más comúnmente.
- Análisis de secuencia del gen, que permite detectar SNPs, así como otras variaciones (microdeleciones e inserciones, mutaciones en los sitios de splicing…).
- Análisis de duplicación o deleción.
Para seleccionar la prueba molecular adecuada, se sigue un orden. En primer lugar, se confirma el diagnóstico molecular preliminar basado en síntomas con pruebas bioquímicas. Seguidamente, se realiza un análisis de mutaciones dirigido. Si no se encuentra ninguna o sólo una mutación causante de la enfermedad, se procede al análisis de secuencia. Si aún es necesario, se puede hacer un análisis de duplicación o deleción para detectar mutaciones específicas menos comunes no incluidas en la primera prueba. Por otro lado, para poder diagnosticar portadores de un alelo mutado, se necesita saber previamente cuál es la mutación causante de la enfermedad en su familia. Aunque estas personas sean asintomáticas, esta prueba puede ser de importancia a la hora de tener hijos.
Posibles complicaciones
El cribado en recién nacidos evita complicaciones potencialmente fatales (morbilidad y mortalidad) durante el periodo neonatal, es decir que la implantación precoz de una dieta restrictiva en galactosa puede salvar la vida y asegurar la casi total integridad funcional de los pacientes. Sin embargo no está demostrado que el diagnóstico y tratamiento de precoz de la enfermedad influya en el desarrollo de complicaciones a largo plazo. A pesar de un tratamiento dietético adecuado desde el momento de su detección, se ha observado que niños con galactosemia siguen teniendo un mayor riesgo de retrasos en el desarrollo, problemas en el habla y anormalidades en la función motora. Asimismo, mujeres con esta enfermedad mantienen un alto riesgo de padecer insuficiencia ovárica prematura. Con los análisis comparados que se han hecho hasta el momento no se ve una clara relación entre las diferentes condiciones de tratamiento, como el inicio del tratamiento y el grado de restricción de la dieta, y las complicaciones a largo plazo. Estas aparecen en proporciones similares independientemente de dichas conclusiones. Los tres tipos de galactosemia, causados cada uno por la mutación de un gen diferente, tienen manifestaciones clínicas diferentes. Tal y como se ha comentado anteriormente, los síndromes clínicos para cada tipo de galactosemia son distintos:
- Deficiencia de GALK: cataratas
- Deficiencia de GALE: gran variabilidad de manifestaciones clínicas
- Deficiencia de GALT: conjunto de manifestaciones clínicas conocidas como galactosemia clásica.
Complicaciones deficiencia GALT
Las personas que sufren galactosemia clásica son las que manifiestan más complicaciones. Esto se debe a que el déficit de GALT impide la adecuada oxidación de la galactosa que se concentra en exceso en el plasma sanguíneo convirtiéndose en galactonato y galactitol. El galactitol no puede ser metabolizado, y la parte que no es eliminada por la orina es la que probablemente tiene importantes efectos tóxicos para el organismo. Las complicaciones de la galactosemia clásica más frecuentes son:
- Complicaciones neurológicas:[8] Las personas con Galactosemia tienen anomalías en la mielinización, tanto en cerebro como en el cerebelo, debido a una afectación en la producción de mielina.
- Retraso mental: la capacidad intelectual de los pacientes con galactosemia tratadas de forma precoz y que siguen bien la dieta está por debajo de la media. Aproximadamente la mitad de los niños con galactosemia tienen una capacidad intelectual en la franja borderline a normal-baja y la otra mitad dentro de la franja de la normalidad. Existe sin embargo una gran variabilidad.
- Afectación del lenguaje: alta prevalencia de la dispraxia verbal.
- Otras: Enlentecimiento de la velocidad de procesamiento, afectación de funciones atencionales, afectación de funciones ejecutiva
- Problemas motrices: dificultades en motricidad amplia y fina, temblor, ataxia, dismetría.
- Cataratas
- Fracaso hepático grave
- Déficit inmunitario con tendencia a sepsis por E. Coli: debido a una inhibición de la actividad bactericida de los leucocitos.
- Fallo ovárico prematuro: la galactosemia tiene efectos ovotóxicos que atenúan FSH.
- Disminución de la densidad ósea: pudiendo llegarse a desarrollar problemas de osteoporosis.
Tratamiento
El tratamiento de la galactosemia también se divide según el tipo de deficiencia enzimática, y se basa principalmente en un estricto control dietético.[9]
- Deficiencia de GALK: debe procederse a la eliminación de la leche de la dieta (parecen tolerarse otras fuentes menores de galactosa).Si el tratamiento es precoz, las cataratas pueden resolverse. Si el diagnóstico es tardío es habitual que las cataratas deben ser operadas.
- Deficiencia de GALE: hay gran variabilidad de manifestaciones clínicas. Algunas formas no precisan tratamiento (solo control). Las formas graves deben ser tratadas con dieta restrictiva en galactosa.
- Deficiencia de GALT: en este caso el tratamiento consiste en eliminar toda administración de la galactosa en la dieta, incluso antes de confirmar el diagnóstico. El tratamiento debe mantenerse durante todo la vida
La ingesta media normal diaria de galactosa es de unos 6,5 g mientras que en pacientes con galactosemia clásica se recomienda una dieta restrictiva que contiene aproximadamente unos 40 mg de galactosa. No se sabe exactamente que cantidad de galactosa pueden ingerir los enfermos de galactosemia por eso debe procurarse una ingesta mínima sin que en ningún caso se superen los 125 mg diarios.
La fuente principal de galactosa de la dieta es la lactosa procedente casi exclusivamente de la leche de mamíferos y de derivados lácteos, aunque también hay lactosa en excipientes y gran variedad de productos manufacturados e industriales.
Se recomienda la administración de leche con proteínas a base de soja. Con la introducción de la alimentación complementaria es algo más complicado mantener una dieta absolutamente libre de galactosa, debido a las dificultades para determinar el contenido real de galactosa libre o ligada de los alimentos.
La dieta de los pacientes con galactosemia tiene con objetivo añadir la menor cantidad posible de galactosa externa (exógena) a la que ya genera el propio organismo (síntesis endógeno).Con este fin se dividen los alimentos en 3 grupos:
- Alimentos que casi no poseen galactosa (<5 mg/100 g)
- Alimentos que deben ser utilizados bajo control (5 mg-20 mg/100 g): fórmulas de soja con harina de soja, calabaza, col de Bruselas, pimientos, puerro, tomate, cacao, levadura, pipas de girasol, sandía, kiwi…
- Alimentos prohibido por su alto contenido en galactosa (>20 mg/100 g): leche y cualquier derivado lácteo, vísceras, guisantes, dátiles, higos secos, pasas…
Aparte de la dieta se tiene que tiene en cuenta dos tratamientos complementarios.
El primero es el suplemento de calcio porqué es habitual que la dieta para niños galactósemicos no asegure las cantidades necesarias de calcio. Las mujeres con déficit de GALT (galactosemia clásica) pueden necesitar tratamiento hormonal para las deficiencias ováricas.
En los 3 tipos de galactosemias es necesario un control del tratamiento y un control general de la evolución de los pacientes. En cada caso dependerá de las características propias de cada una de las 3 deficiencias y de las necesidades individuales. Dada la magnitud de las complicaciones y del tratamiento a seguir, dichos controles son más exhaustivos y frecuentes en el caso de la deficiencia GALT (galactosemia clásica).
Véase también
Referencias
- Perry, A. Frey (marzo de 1996). «The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose». The FASEB Journal. vol. 10 (4): 461-470. PMID 8647345. Consultado el 25 de noviembre de 2012.
- Holden, Hazel M.; Ivan Rayment and James B. Thoden (agosto de 2003). «Structure and Function of Enzymes of the Leloir Pathway for Galactose Metabolism». The Journal of Biological Chemistry. Vol. 278 (45): 43885-43888. doi:10.1074/jbc.R300025200. Consultado el 25 de noviembre de 2012.
- «GALK1» (en inglés). 19 de noviembre de 2012. Consultado el 25 de noviembre de 2012.
- Diagnóstico y tratamiento de enfermedades metabólicas. Moreno, B. 1.ª edición. 1997 Editorial Díaz de Santos S.A. ISBN 978-84-7978-303-7.
- «GALE» (en inglés). 19 de noviembre de 2012. Consultado el 25 de noviembre de 2012.
- «GALT» (en inglés). 19 de noviembre de 2012. Consultado el 25 de noviembre de 2012.
- «Florida Health Finder».
- «complicaciones neurológicas de la galactosemia clásica».
- http://www.ae3com.eu/protocolos/protocolo7.pdf
Enlaces externos
- Insights into the pathogenesis of galactosemia.
- Natural Course and Treatment of Neuropsychological Deficits in a Child with Early-treated Galactosemia. (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).
- Parents of Galactosemic Children, Inc.
- Asociación Española para la Galactosemia http://www.galactosemia.es
- Facebook Asociación Española para la Galactosemia https://www.facebook.com/AEGalactosemia
- A common mutation associated with the Duarte galactosemia allele.
- Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene.
- ARHI: A new target of galactose toxicity in Classic Galactosemia.
- FloridaHealthFinder.
- Galactosemia por Genetics Home Reference.
- Vía de Leloir.
- The Leloir pathway.
- Galactosemia.
- Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa.
- Revisión completa de la enfermedad y su diagnóstico clínico en GeneTests (NCBI)
- Página de la enfermedad en OMIM
| Control de autoridades |
|
|---|
 Datos: Q774483
Datos: Q774483 Multimedia: Galactosemia / Q774483
Multimedia: Galactosemia / Q774483