Hibridación in situ
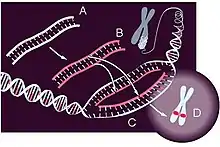
La hibridación in situ (ISH) es una técnica que se utiliza para la localización y la detección de secuencias de ADN y de ARN específicas en las células, cromosomas o tejidos preservados,[1] mediante la formación de una molécula híbrida entre una molécula endógena de ARN o ADN de la célula y una sonda complementaria de ARN o de ADN monocatenario.[2] La técnica de hibridación in situ se basa en la capacidad que poseen los ácidos nucleicos para hibridarse entre sí, es decir, la existencia de determinada secuencia de ADN o ARN, que resulta complementaria con otra secuencia.[3] Su utilidad reside en la capacidad de poder demostrar mediante la utilización de una sonda (formada por una secuencia de ADN previamente conocida) marcada con un isótopo radiactivo, la presencia de determinada secuencia de ADN o ARN complementaria, en la muestra a estudiar. Cuando una de las dos hebras está marcado, los híbridos reconocidos pueden ser detectados por varios métodos, que pueden ser fluorescente y no fluorescente.[1] La detección in situ permite visualizar de forma directa la ubicación espacial de secuencias específicas, como se observa en la Figura 1, lo cual es esencial para dilucidar la organización y función génica, debido a esto esta técnica es de gran importancia en diversos campos, entre los cuales se encuentra el diagnóstico de rearreglos cromosomales, la detección de infecciones virales y el análisis de la función génica durante el desarrollo embrionario.
Historia
La técnica se desarrolló en el año de 1969 por dos grupos de investigación, Gall y Pardue e independientemente por Jonh et al, en el mismo año. Gall y colaboradores describieron una técnica en la cual se formaban híbridos moleculares entre RNA y DNA. La técnica fue llevada a cabo en ovocitos de Xenopus laevis, mediante la hibridación de rRNA con rDNA extracromosomal; en los cuales detectaron la localización diferencial de los híbridos de ácidos nucléicos a distintos estadios usando microautoradiografia.[3] Esta técnica se caracterizó por permitir que la secuencia de ácidos nucleicos fuera detectada dentro de la célula sin alterar la morfología celular o la integridad de sus compartimentos. El uso de Hibridación in situ para "contar e identificar organismos" fue propuesto por Olsen,[4] y luego introducida a la bacteriología en el año 1988 por el grupo de Giovannoni, quienes fueron los primeros en utilizar sondas de oligonucleótidos marcadas radioactivamente y dirigidas al ARNr, para la detección microscópica de bacterias.[5]
Fundamento
El objetivo de la hibridación in situ es determinar la presencia o ausencia de secuencias de DNA o RNA de interés, así como localizar estas secuencias en células específicas o sitios cromosómicos.[6] Condiciones óptimas para la hibridación de extractos purificados de DNA o RNA en soportes sólidos han sido arduamente estudiadas. Con el uso de muestras tisulares y celulares; sin embargo, nuevos problemas tales como la unión no específica de las sondas han sido encontrados. La sensibilidad de la hibridación in situ depende de las siguientes variables: la preparación de la muestra (tejido o célula), construcción de la sonda, etiquetado de la sonda y la sensibilidad del método usado para la detección.[7]
- Preparación de la muestra: Un pretratamiento puede ser llevado a cabo antes de hibridación para aumentar la eficiencia y reducir la tinción no específica. El tratamiento con proteasas (proteinasa K la más común) facilita el acceso hacia la secuencia objetivo. La acetilación con 0,25% de anhídrido acético o 0.1 M trietanolamina reduce la unión de las sondas en el tejido. La optimización de la preparación de la muestra, incluido la fijación y el almacenamiento, es importante para la detección intracelular de ácidos nucleicos.[8] La fijación necesita preservar el DNA o RNA y la morfología del tejido. En contraste, los procesos de síntesis y degradación enzimática de DNA o RNA deben mantenerse estables, por lo tanto el promedio de vida del mRNA es un factor importante. En este contexto, el tejido a ser fijado debe congelarse tan pronto sea posible tras su escisión, el tiempo entre la escisión y fijación se toma en cuenta al momento de interpretar los resultados. Se utiliza para-formaldehído para la fijación, en algunos casos seguido por la adición de glutaraldehido. La fijación de ciertos tejidos se ve favorecida por mezclas de ácido acético-alcohol o el fijador de Bouin.[7]
- Construcción de la sonda: Diferentes tipos de sondas tales como cDNA, cRNA y oligonucleótidos sintéticos se utilizan para la hibridación in situ. La elección de la sonda dependerá de la sensibilidad y especificidad, facilidad de producción, facilidad de la sonda para penetrar el tejido, aplicación y la reproducibilidad del método. El tamaño óptimo de la sonda es 50-300 nucleótidos.[6]
- Etiquetado de la sonda: El etiquetado de la sonda puede realizarse mediante la incorporación de radioisótopos ( 3H, 32P, 35S, 14C, 125I ) o marcadores no radioactivos (biotina, anticuerpos específicos, entre otros). El uso de marcadores radioactivos se considera el método más sensible. Los sitios de hibridación pueden visualizarse mediante radiografía con rayos-X o emulsiones líquidas.[7]
- Sensibilidad del método: en función de la sonda utilizada y la secuencia de interés distintas reacciones de control pueden llevarse a cabo tales como Northern blot o Southern blot, Inmunoquimica, hibridación con fragmentos de secuencia conocida, entre otros.[7]
Ventajas y Desventajas de la Hibridación in situ
La principal ventaja de la técnica es que permite usar al máximo la muestra de un tejido, logrando realizar cientos de hibridaciones diferentes en el mismo tejido.[1] Sin embargo la técnica de hibridación in situ presenta la desventaja de estar limitada por su incapacidad para detectar secuencias que tienen bajo número de copias de ADN y ARN. Debido a esto se han desarrollado varias estrategias para mejorar la sensibilidad de la hibridación in situ por amplificación de cualquiera de las secuencias de ácido nucleico antes de la ISH o detección de la señal después de que se complete la hibridación.[9]
Tipos de Hibridación in situ
Desde la aparición de la hibridación in situ, numerosos métodos han sido desarrollados. Entre los métodos más comunes tenemos:[1]
PCR con hibridación in situ
Se usa para la amplificación de secuencias específicas de ADN o ARN en células o secciones de tejido, de forma que el número de copias se incrementen en PCR in situ a niveles detectables por métodos estándar de hibridación in situ. Su principal utilidad se da para identificar secuencias de ADN que no son fáciles de detectar usando hibridación in situ estándar. Sin embargo esta técnica posee una baja eficiencia de amplificación y una mala reproducibilidad.[10] PCR in situ directo suele resultar en falsos positivos debido a la incorporación de oligonucleótidos no marcados específicos en fragmentos de DNA. Estos artefactos exhiben señales nucleares y son evidentes en células apoptóticas. Los artefactos resultantes de la técnica se reducen por la acción de una exonucleasa o por reparación de estos fragmentos de DNA con T4 DNA ligasa o mediante el uso de ciclos con sondas marcadas. Los falsos positivos no se eliminan por completo pero, el grado de detección específica aumenta.
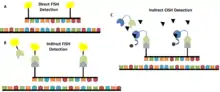
Los métodos indirectos proveen máxima especificidad. Usualmente se usan sondas largas completas o sondas genómicas que incrementan el número de moléculas reporteras para obtener una buena señal. La naturaleza de los sistemas reporteros influye en la especificidad de la técnica. Las sondas radioactivas presentan alta estabilidad, sin embargo, son muy costosas. Los sistemas colorimétricos basados en fosfatasa alcalina o peróxidos resultan en una señal pobre. Los sistemas de detección basados en fluorescencia no son recomendables dado que no conservan la morfología del tejido.[11]
.jpg.webp)
Hibridación in situ con fluorescencia (FISH)
Es una técnica que detecta secuencias de ácidos nucleicos en células o tejidos preservados mediante el empleo de una sonda marcada con un fluorocromo, la cual va dirigida hacia un lugar específico del cromosoma y que emite fluorescencia que puede ser observada por medio de un microscopio.[12] Se caracteriza por permitir la visualización directa de las alteraciones genéticas en la célula.[1] El principio básico de FISH es hibridar una sonda de oligonucleótidos marcada con fluorescencia a una secuencia de interés dentro de un microorganismo y visualizar o contar la señal fluorescente resultante por microscopia u otro método.
El procedimiento básico FISH incluye: 1) fijación y permeabilización; 2) hibridación; 3) lavado; y 4) la microscopía (para el recuento y visualización) o citometría de flujo (para el recuento de alta velocidad). En ciertos tipos de muestras el procedimiento FISH puede ir precedido de un aislamiento de células o método de concentración. Este principio básico y los pasos implicados en la FISH se muestran en la Figura 1 y se describen en detalle a continuación. Con independencia del enfoque aplicado FISH específica, sólo las células que contienen el ADN diana son reconocidos por la sonda, y estarán etiquetados con fluorescencia cuando se visualizan con las técnicas adecuadas.[13]
- La fijación y permeabilización. Este paso permite preservar la integridad y la forma de todas las células, además el mismo facilita la penetración de las sondas FISH fluorescente en la célula y protege el gen diana de la degradación durante el almacenamiento y el análisis. El proceso consiste en cubrir la muestra con un agente de fijación; normalmente se usa como agente de fijación el formaldehído y etanol. El agente de fijación tiene la función de permeabilizar las células para permitir la entrada de oligonucleótidos marcados a las células y la posterior difusión de las sondas a sus dianas intracelulares. Luego de la fijación se elimina el fijador residual y se transfiere la muestra a un portaobjetos de microscopio.
- La hibridación. Durante la hibridación una sonda marcada con fluorescencia se une a la secuencia de interés. Sólo organismos que contienen los genes diana incorporan el marcador fluorescente, de modo que se pueden visualizar usando un microscopio u otra técnica. Otros tipos de técnicas de marcaje de células que se utilizan con menos frecuencia que las sondas de oligonucleótidos incluyen combinaciones de moléculas indicadoras (digoxigenina), anticuerpos fluorescentes y la amplificación de la señal enzimática (tryamide), o sondas de polirribonucleótidos.
- Lavado. Durante la etapa de lavado los portaobjetos de microscopio que contienen las células fijadas e hibridadas se enjuagan brevemente para eliminar la sonda no unida que pudiera interferir con la cuantificación de los microorganismos diana. Los portaobjetos se secan a continuación, y se cubren con un agente de antidecoloramiento.
- La visualización o conteo. Células híbridas se visualizan por microscopía de epifluorescencia o por citometría de flujo. Miscroscopia de fluorescencia utiliza una luz de alta intensidad para iluminar una muestra, que excita las especies fluorescentes, generando una imagen de las moléculas fluorescentes unidas a las sondas de oligonucleótidos. En esta técnica las células son enumeradas por el técnico de laboratorio o por un conteo programas automatizados. Sin embargo un conteo más eficiente de células marcadas se consigue con citometría de flujo, en la cual se diluyen las células marcadas de manera que las células individuales pasan a través de un rayo láser, que detecta y cuenta células marcadas fluorescentemente.
Multicolor FISH
En el FISH multicolor, se usa dos o más sondas marcadas específicamente, las cuales luego de hibridarse con las secuencias de la muestra, son identificadas con diferentes colores fluorescentes. Esta es una técnica eficaz para buscar anomalías cromosómicas, que a pesar de ser cara, presenta la ventaja de permitir pueden evaluar múltiples sitios cromosómicos.[1] Los tipos de FISH multicolor incluyen FISH multiplex, cariotipaje espectral, bandas de color entre especies (cross-species color banding) e hibridación genómica comparativa, estas técnicas se describen a continuación:
En el multiplex-FISH, el cariotipo espectral del genoma humano se realiza con 24 colores diferentes. La técnica de multiplex-FISH permite la detección de muchos cambios cromosómicos en el genoma en una sola reacción de hibridación y es considerado un método fiable para el diagnóstico de anomalías cromosómicas. Los dos sistemas de multiplexado-FISH y cariotipo espectral tienen diferentes maneras de adquirir y procesar imágenes de cromosomas, pero ambos proporcionan la misma información.
Otro tipo de FISH Multicolor es cross-species color banding, el cual combina la sensibilidad del método tradicional de bandas G con la objetividad y la eficiencia de una técnica molecular. Este método utiliza una sonda procedente de una especie de mono, el gibón, para examinar los cromosomas humanos.
La hibridación genómica comparativa es un buen método para investigar la composición genética de las células a partir de dos fuentes diferentes. Este enfoque se utiliza para la investigación de las variaciones en el número de copias de ADN y se caracteriza por su capacidad de revelar alteraciones genéticas en todas las áreas del genoma en un experimento por FISH y análisis de imágenes por computadora.[1]
Hibridación in situ con cromógenos (CISH)
En este tipo de hibridación se utilizan reacciones de peroxidasa o fosfatasa alcalina usando microscopía de campo brillante en tejidos fijados por formalina y embebidos en parafina. En esta técnica los anticuerpos se conjugan con una enzima que cataliza reacciones de sustratos cromogénicos.[1] Los cromógenos resultantes precipitan en el lugar de destino de la sonda y se pueden detectar con un microscopio de campo brillante estándar.
Esta técnica además de proporcionar una alternativa económica fácil de usar para FISH, presenta la ventaja de que permite la visualización del tejido, incluyendo el núcleo y forma de la célula, junto con la señal de la sonda en la misma imagen. A diferencia de la mayoría de los reactivos de detección fluorescentes, agentes cromogénicos utilizados en la mayoría de los métodos de CISH son químicamente estables y no se desvanecen con el tiempo, lo que permite un fácil almacenamiento y posibilita reexaminar las muestras.[14] En las Figura 3 se observa las diferencias entre la hibridación in situ fluorescente FISH y l la hibridación in situ con cromógenos (CISH). Entre las aplicaciones de CISH tenemos que la técnica permite examinar la deleción de un gen, translocaciones cromosómicas, y el número cromosómico.
Hibridación in situ genómica (GISH)
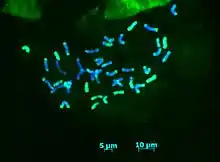
Se utiliza con el fin de distinguir los cromosomas de diferentes progenitores o de diferentes genomas en híbridos interespecíficos o alopoliploides. La característica principal de esta técnica es que mientras FISH utiliza secuencias específicas de ADN como sondas, GISH utiliza ADN genómico de una especie como sonda. La técnica fue desarrollada inicialmente para líneas celulares híbridas de animales (1986) y, tiempo después para plantas a cargo del Instituto de Fitomejoramiento, Cambridge (1987), donde obtuvo su nombre. La técnica involucra la extracción, seguido por el etiquetado de DNA con un isótopo radiactivo del organismo que se utiliza como sonda para identificar el genoma de interés. Las secciones del genoma que son similares a la sonda se hibridan y forman un complejo, el cual ahora se encuentra etiquetado. Las partes restantes del genoma sin hibridar son teñidas para su visualización.
El gran avance de esta técnica en relación con FISH es el etiquetado de genomas enteros, haciéndolo ventajoso en muchos estudios, por ejemplo, en la identificación de genomas y en el análisis meiótico.[15] Un ejemplo de la técnica de GISH se observa en la figura 4 donde se visualiza la hibridación genómica in situ (GISH) en cromosomas Thinopyrum intermedium.
Véase también
Referencias
- Jensen, Ellen (1 de agosto de 2014). «Technical Review: In Situ Hybridization». The Anatomical Record (en inglés) 297 (8): 1349-1353. ISSN 1932-8494. doi:10.1002/ar.22944. Consultado el 2 de diciembre de 2016.
- Gall, Joseph G. (1 de abril de 2016). «The origin of in situ hybridization - A personal history». Methods (San Diego, Calif.) 98: 4-9. ISSN 1095-9130. PMC 4808352. PMID 26655524. doi:10.1016/j.ymeth.2015.11.026. Consultado el 2 de diciembre de 2016.
- Ramos, Blanca. HIBRIDACIÓN in situ.
- Olsen, G. J.; Lane, D. J.; Giovannoni, S. J.; Pace, N. R.; Stahl, D. A. (1 de enero de 1986). «Microbial ecology and evolution: a ribosomal RNA approach». Annual Review of Microbiology 40: 337-365. ISSN 0066-4227. PMID 2430518. doi:10.1146/annurev.mi.40.100186.002005. Consultado el 2 de diciembre de 2016.
- Giovannoni, S J; DeLong, E F; Olsen, G J; Pace, N R (1 de febrero de 1988). «Phylogenetic group-specific oligodeoxynucleotide probes for identification of single microbial cells.». Journal of Bacteriology 170 (2): 720-726. ISSN 0021-9193. PMC 210714. PMID 2448289. Consultado el 2 de diciembre de 2016.
- Jensen, Ellen (1 de agosto de 2014). «Technical Review: In Situ Hybridization». The Anatomical Record (en inglés) 297 (8): 1349-1353. ISSN 1932-8494. doi:10.1002/ar.22944. Consultado el 4 de diciembre de 2016.
- Höfler, Heinz (1 de junio de 1987). «What's New in “in Situ Hybridization”». Pathology - Research and Practice 182 (3): 421-430. doi:10.1016/S0344-0338(87)80080-8. Consultado el 4 de diciembre de 2016.
- Egger, D.; Troxler, M.; Bienz, K. (1 de junio de 1994). «Light and electron microscopic in situ hybridization: non-radioactive labeling and detection, double hybridization, and combined hybridization-immunocytochemistry». The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society 42 (6): 815-822. ISSN 0022-1554. PMID 8189042. Consultado el 4 de diciembre de 2016.
- «Recent Developments in Signal Amplification Methods for In S... : Diagnostic Molecular Pathology». LWW. Consultado el 4 de diciembre de 2016.
- Qian, Xiang; Lloyd, Ricardo V. (1 de marzo de 2003). «Recent developments in signal amplification methods for in situ hybridization». Diagnostic Molecular Pathology: The American Journal of Surgical Pathology, Part B 12 (1): 1-13. ISSN 1052-9551. PMID 12605030. Consultado el 4 de diciembre de 2016.
- PRINS and In Situ PCR Protocols | Franck Pellestor | Springer (en inglés). Consultado el 25 de enero de 2017.
- Martínez, Raúl Rodríguez (1 de enero de 2013). «Aplicaciones e inconvenientes de la técnica Hibridación in situ Fluorescente (FISH) en la identificación de microorganismos». Salud Uninorte. Consultado el 4 de diciembre de 2016.
- «9 Fluorescence In Situ Hybridization (FISH)». www.itrcweb.org (en inglés estadounidense). Consultado el 20 de enero de 2017.
- 617feng,厦门百邑科技. «CISH/SISH - CytoTest». www.cytotest.com. Archivado desde el original el 2 de febrero de 2017. Consultado el 21 de enero de 2017.
- «Genomic in situ hybridization in plants». GMR | Genetics and Molecular Research. 24 de agosto de 2015. Consultado el 4 de diciembre de 2016.
 Datos: Q2304142
Datos: Q2304142 Multimedia: In situ hybridization / Q2304142
Multimedia: In situ hybridization / Q2304142