Histiocitosis de células de Langerhans
La histiocitosis de células de Langerhans (HCL) (también conocida como enfermedad de Abt-Letterer-Siwe, Histiocitosis X y enfermedad de Hand-Schüller-Christian) es una enfermedad rara que involucra la proliferación anormal de células de Langerhans, células anormales que provienen de la médula ósea y capaces de migrar desde la piel hasta los linfonodos. Clínicamente las manifestaciones pueden variar desde lesiones óseas aisladas hasta una enfermedad multisistémica.
| Histiocitosis de células de Langerhans | ||
|---|---|---|
 | ||
| Especialidad | oncología | |
| Sinónimos | ||
| ||
HCL es parte de un grupo de enfermedades sindromáticas llamadas histiocitosis, las cuales se caracterizan por la proliferación anormal de histiocitos (un antiguo término que hace alusión a las células dendríticas activadas y a los macrófagos).
Estas enfermedades se relacionan con otras formas de proliferación anormal de células blancas, tales como la leucemia y los linfomas. La enfermedad ha tenido muchos nombres incluyendo Hand-Schuller-Christian, Abt-Letterer-Siwe e Histiocitosis X, hasta que finalmente fue renombrada en 1985 por la sociedad de histiocitosis.[1]
Es un padecimiento de la niñez, no familiar y que no está restringido a alguna población. Clínicamente provoca diabetes insípida. Otras manifestaciones de esta enfermedad pueden ser exoftalmos, eccema o dermatitis eccematosa, otitis media e infecciones del sistema respiratorio alto. Los ganglios linfáticos, el hígado y el bazo pueden estar aumentados de tamaño.
Epidemiología
La HCL usualmente afecta a los niños entre 1 y 15 años, con un pico de incidencia entre 5 y 10 años. Entre los niños bajo 10 años, la incidencia anual es de 1 de cada 200 000 niños en Estados Unidos[2] y en adultos es bastante más rara, un caso cada 560 000.[3] Es mucho más rara en adultos.[4]
En un estudio epidemiológico de casos y controles, realizado en Los Ángeles, Cal. Venkatramani et al, encontraron que la población predominante con HCL era de origen hispano y no caucásica (53 % vs 18 %), por lo que hay que considerar que estudios donde inferían una mayor incidencia en raza caucásica estaba relacionada por el tipo de población estudiada. En este estudio tampoco hubo predominancia del sexo masculino, 43 % fueron masculinos y 57 % femeninos (Venkatramani R, 2012).
Las manifestaciones pulmonares de la histiocitosis X se caracterizan por darse en jóvenes, fumadores y sobre todo, distribuidas las lesiones en los lóbulos superiores de ambos pulmones.[5]
La HCL es usualmente esporádica sin relación hereditaria pero se ha encontrado cierto patrón familiar en un número limitado de casos. La enfermedad de Hashimoto-Pritzker es una variante congénita de la enfermedad de la HCL crónica diseminada.[6]
Clasificación
Existen muchas antiguas clasificaciones, actualmente se reconocen tres formas de HCL:[7]
| Clasificación anterior | Clasificación actual |
|---|---|
| 1. Granuloma Eosinófilo | HCL Crónica Focal |
| 2. Hand-Schuller-Christian | HCL Crónica diseminada |
| 3. Letterer-Siwe | HCL Aguda Diseminada |
Clínica
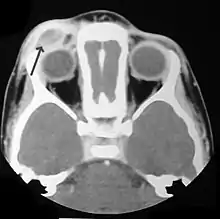
La HCL provoca una respuesta inflamatoria inespecífica, la cual incluye fiebre, letargia y pérdida de peso. Los órganos afectados darán síntomas y signos más específicos.
- Hueso. El síntoma más frecuentemente visto ya sea en la variante unifocal o multifocal de la enfermedad, es el edema y dolor óseo. El cráneo es el hueso que con mayor frecuencia se ve afectado, seguido por los huesos largos de las extremidades superiores y los huesos planos. La infiltración en las manos y pies resulta poco común. Las lesiones son de tipo osteolítica lo cual puede producir fracturas en hueso patológico.
- Piel: Generalmente se manifiesta como rash el cual varía desde lesiones eritematosas hasta pápulas pronunciadas en las áreas de los pliegues. Hasta el 80 % de los pacientes con HCL tienen lesiones extensivas y eruptivas en el cuero cabelludo.
- Médula ósea: La pancitopenia con sobre-infecciones generalmente implican un peor pronóstico. La anemia puede ser debida a un número de factores que no necesariamente implican infiltración a nivel de la médula ósea.
- Linfonodos: La hepatomegalia (20 % de los pacientes), esplenomegalia (30 % de los pacientes) y linfoadenopatías (50 % de los pacientes) son característicos del cuadro clínico.[8]
- Glándulas endocrinas: El eje hipotálamo-hipófisis comúnmente está involucrado. La diabetes insípida es la más afectada. El déficit hormonal a nivel de la neurohipófisis es permanente.
- Pulmón: en algunos pacientes que son asintomáticos, el diagnóstico se realiza accidentalmente al encontrar nódulos pulmonares en la radiografía de tórax. En otros casos se observa, tos crónica y disnea.
- Menos frecuentemente se puede ver afectado el sistema nervioso y el aparato gastrointestinal.
Diagnóstico
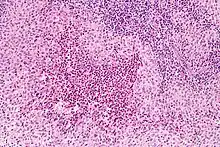
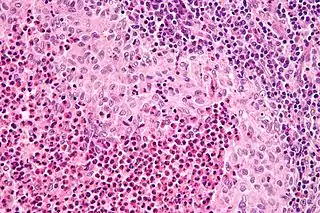
El diagnóstico se realiza por confirmación histológica a través de una biopsia tisular. La tinción de hematoxilina-eosina de la biopsia mostrará algunas características de las células de Langerhans como por ejemplo, gránulos plasmáticos rosados y margen celular distintivo. La presencia de gránulos de Birbeck en la microscopía electrónica y las características inmunohistoquímicas como por ejemplo la positividad para CD1, resultan pruebas más específicas. Algunos exámenes de laboratorio de rutina como el hemograma, perfil hepático, y otras pruebas sirven para orientar el diagnóstico (principalmente para descartar otras posibles causas). Los exámenes imagenológicos mostrarán lesiones osteolíticas y daño pulmonar. Esto último se puede evidenciar en la radiografía de tórax al mostrar lesiones micronodulares e infiltrados pulmonares en las zonas bajas y medias. La resonancia nuclear magnética o la tomografía axial computarizada puede mostrar infiltración a nivel de la silla turca. La medición del eje endocrino y la biopsia de la médula ósea también pueden resultar útiles para el diagnóstico.
Tratamiento
El tratamiento está indicado para la variante difusa de la enfermedad. Las lesiones óseas solitarias pueden ser manejadas a través de la escisión de la lesión o radioterapia local. Sin embargo, la enfermedad sistémica, generalmente requiere quimioterapia. El uso de corticoides sistémicos solos o en coterapia con quimioterapia, es la forma de manejo más habitual. Cremas con corticoides locales pueden ser aplicados para las lesiones en la piel. La deficiencia endocrina generalmente requiere suplementos de por vida, por ejemplo, desmopresina en el caso de diabetes insípida la cual puede ser incluso aplicada vía nasal. Los agentes quimioterapéuticos como por ejemplo los alquilantes, antimetabolitos y los alcaloides de la Vinca ya sea en monoterapia o en terapia combinada pueden llevar a la remisión de la enfermedad difusa.
Pronóstico
El pronóstico en la enfermedad focal es muy bueno. En el caso de la enfermedad sistémica, un 60 % tienen un curso crónico, mientras un 31 % remite y un 10 % muere.
Enlaces externos
Referencias
- «Histiocytosis syndromes in children. Writing Group of the Histiocyte Society». Lancet 1 (8526): 208-9. 1987. PMID 2880029. doi:10.1016/S0140-6736(02)95528-5.
- «MedlinePlus Medical Encyclopedia: Histiocytosis». Consultado el 10 de mayo de 2007.
- «Histiocytosis Association of Canada». Archivado desde el original el 14 de mayo de 2007. Consultado el 16 de mayo de 2007.
- Gerlach B, Stein A, Fischer R, Wozel G, Dittert D, Richter G (1998). «[Langerhans cell histiocytosis in the elderly]». Der Hautarzt; Zeitschrift für Dermatologie, Venerologie, und verwandte Gebiete (en alemán) 49 (1): 23-30. PMID 9522189.
- «Diagnóstico y tratamiento de HISTIOCITOSIS DE CÉLULAS DE LANGERHANS en edad pediátrica».
- Kapur P, Erickson C, Rakheja D, Carder K, Hoang M (2007). «Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center». J. Am. Acad. Dermatol. 56 (2): 290-4. PMID 17224372. doi:10.1016/j.jaad.2006.09.001.
- http://www.actaodontologica.com/ediciones/2005/2/histiocitosis_celulas_langerhans_cronica_focal.asp
- «Langerhans cell histiocytosis - Patient UK». Consultado el 10 de mayo de 2007.
| Control de autoridades |
|
|---|
 Datos: Q374036
Datos: Q374036 Multimedia: Langerhans cell histiocytosis / Q374036
Multimedia: Langerhans cell histiocytosis / Q374036