Holoprosencefalia
La holoprosencefalia constituye un amplio espectro de malformaciones del cráneo y la cara debidas a una anormalidad compleja del desarrollo del cerebro que tienen en común la ausencia del desarrollo del prosencéfalo, que es el lóbulo frontal del cerebro del embrión.[2] Durante el desarrollo normal se forma el lóbulo frontal y la cara comienza a desarrollarse en la quinta y sexta semana del embarazo. La holoprosencefalia es causada por la falta de división del lóbulo frontal del cerebro del embrión para formar los hemisferios cerebrales bilaterales (las mitades izquierda y derecha del cerebro), causando defectos en el desarrollo de la cara y en la estructura y el funcionamiento del cerebro.[3]
| Holoprosencefalia o cebocefalia | ||
|---|---|---|
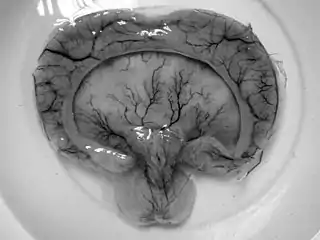 Holoprosencefalia alobar, producto de una madre de 19 años en Tailandia con diabetes gestacional.[1] | ||
| Especialidad | genética médica | |
Causas
Aunque las causas de la mayoría de los casos de holoprosencefalia son teratogénicos, aproximadamente la mitad de todos los casos se deben a causas cromosómicas. Las anomalías cromosómicas, tales como el síndrome de Patau (trisomía 13), el síndrome de Edwards (trisomía 18), el síndrome de Meckel-Gruber y ciertas triploidías se han podido asociar con la holoprosencefalia.[2] Los hijos de madres diabéticas y la ingesta elevada de alcohol en esta parte del desarrollo embrionario (ya que destruye las células de la línea media de estructuras craneofaciales) tienen un riesgo mayor de padecer el trastorno.
Clasificación
Existen tres clases de holoprosencefalia, basados en la separación del prosencéfalo:[2]
- Holoprosencefalia alobar es el tipo más grave, en la cual el cerebro no logra separarse y se asocia generalmente a anomalías faciales severas (fusión de los ojos, anomalías del tabique nasal.)[4]
- Holoprosencefalia semilobar, en la cual los hemisferios del cerebro tienen una leve tendencia a separarse, constituye una forma intermedia de la enfermedad.[3]
- Holoprosencefalia lobar, en la cual existe una evidencia considerable de separación de los hemisferios del cerebro, es la forma menos grave.[3] En algunos casos de holoprosencefalia lobar, el cerebro del paciente puede ser casi normal.
Cuadro clínico
La holoprosencefalia, denominada anteriormente como anencefalia, consiste en una gama de defectos o malformaciones del cerebro y de la cara. En el extremo más grave de este espectro se encuentran los casos que involucran malformaciones serias del cerebro, malformaciones tan graves que son incompatibles con la vida y a menudo causan la muerte intrauterina espontánea. En el otro extremo del espectro están los individuos con los defectos faciales-que pueden afectar los ojos, la nariz y el labio superior-y el desarrollo normal o casi normal del cerebro.[3] Pueden ocurrir convulsiones o discapacidad intelectual.
El más grave de los defectos (o anomalías) faciales es la ciclopía, caracterizado por el desarrollo de un solo ojo, que se ubica generalmente en el área ocupada normalmente por la raíz de la nariz, y la ausencia de la nariz o una nariz en la forma de una probóscide (un apéndice tubular) situada por encima del ojo.[4]
La etmocefalia es la anomalía facial menos común. Consiste en una probóscide que separa ojos muy juntos, ausencia de la nariz y microftalmía (tamaño anormalmente pequeño de uno o ambos ojos).
La cebocefalia es otra anomalía facial caracterizada por una nariz pequeña y aplastada con un solo orificio nasal situada debajo de unos ojos subdesarrollados y muy juntos.
La anomalía facial menos grave es el labio leporino, también llamado agenesia premaxilar.
Tratamiento
No existe tratamiento para la holoprosencefalia y el pronóstico para los individuos que la padecen es pobre. La mayoría de los que sobreviven no muestran signos de desarrollo significativos. Para los niños que sobreviven, el tratamiento es sintomático (es decir, alivia sólo los síntomas y no las causas del trastorno). Es posible que una mejora en el monitoreo de embarazos de madres diabéticas pueda ayudar a prevenir la holoprosencefalia. No obstante, no existen medios de prevención primaria.
Véase también
Referencias
- Patou Tantbirojn, Mana Taweevisit, Suchila Sritippayawan y Boonchai Uerpairojkit. Diabetic fetopathy associated with bilateral adrenal hyperplasia and ambiguous genitalia: a case report (en inglés). Journal of Medical Case Reports 2008, 2:251doi:10.1186/1752-1947-2-251. Último acceso 8 de enero de 2009.
- BARRIGA OROPEZA, Jorge, MURILLO SANCHEZ, Consuelo, AGREDA GUERRERO, Julio et al. Holoprosencefalia: A propósito de dos casos (en español). Rev. bol. ped. [online]. jan. 2004, vol.43, no.1 [citado 08 Janeiro 2010], p.23-25. ISSN 1024-0675.
- M. Izquierdo, A. Avellaneda (Diciembre de 2003). «Holoprosencefalia». Instituto de Investigación de Enfermedades Raras. Archivado desde el original el 25 de diciembre de 2009. Consultado el 8 de enero de 2010.
- SALDARRIAGA, Wilmar; ISAZA, Carolina; MASTROIACOVO, Pierpaolo and CASTILLA, Eduardo E. Ciclopía en el Hospital Universitario del Valle (Cali, Colombia): reporte de cuatro casos nacidos y revisión de la literatura (en español). Rev Colomb Obstet Ginecol [online]. 2007, vol.58, n.1 [cited 2010-01-08], pp. 70-77. ISSN 0034-7434.
- NINDS: Artículo publicado bajo dominio público.
| Control de autoridades |
|
|---|
 Datos: Q1459821
Datos: Q1459821 Multimedia: Holoprosencephaly / Q1459821
Multimedia: Holoprosencephaly / Q1459821