Mapeo de asociación
El mapeo de asociación (genética), también conocido como "mapeo de desequilibrio de ligamiento", es un método de mapeo de loci de rasgos cuantitativos (QTL) que aprovecha el desequilibrio de ligamiento histórico para vincular fenotipos (características observables) a genotipos (la constitución genética de organismos), descubriendo asociaciones genéticas.
Teoría
El mapeo de asociación se basa en la idea de que los rasgos que han ingresado a una población recientemente solo estarán vinculados a la secuencia genética circundante del ancestro evolutivo original, o en otras palabras, se encontrarán con mayor frecuencia dentro de un haplotipo dado, que fuera de él. Con mayor frecuencia se realiza escaneando todo el genoma en busca de asociaciones significativas entre un panel de SNP (que, en muchos casos, se ven en diapositivas de vidrio para crear "chips SNP") y un fenotipo particular. Estas asociaciones deben verificarse de forma independiente para demostrar que:
- contribuyen directamente al rasgo de interés, o
- están vinculadas a/en desequilibrio de enlace con un locus de rasgo cuantitativo (QTL) que contribuye al rasgo de interés.[1]
El mapeo de asociación busca identificar variantes genéticas funcionales específicas (loci, alelos) vinculadas a diferencias fenotípicas en un rasgo para facilitar la detección del rasgo que causa polimorfismos de secuencia de ADN y la selección de genotipos que se parecen mucho al fenotipo. Para identificar estas variantes funcionales, se requieren marcadores de alto rendimiento como polimorfismos de un solo nucleótido (SNP).[2]
Uso
La ventaja del mapeo de asociación es que puede mapear rasgos cuantitativos con alta resolución de una manera estadísticamente muy poderosa. Sin embargo, el mapeo de asociación también requiere un amplio conocimiento de los SNP dentro del genoma del organismo de interés y, por lo tanto, es difícil de realizar en especies que no han sido bien estudiadas o que no tienen genomas bien anotados.[3] El mapeo de asociación se ha aplicado más ampliamente al estudio de enfermedades humanas, específicamente en forma de un estudio de asociación de genoma completo (GWAS). Se realiza un estudio de asociación de todo el genoma escaneando un genoma completo en busca de SNP asociados con un rasgo de interés particular, o en el caso de una enfermedad humana, con una enfermedad de interés particular.[1][4] Hasta la fecha, se han realizado miles de estudios de asociaciones de genoma amplio en el genoma humano en un intento de identificar los SNP asociados con una amplia variedad de enfermedades humanas complejas (por ejemplo, cáncer, enfermedad de Alzheimer y obesidad). Los resultados de todos estos GWAS publicados se mantienen en una base de datos NIH (figura 1). Sin embargo, si estos estudios han sido clínicamente y/o terapéuticamente útiles, sigue siendo controvertido.
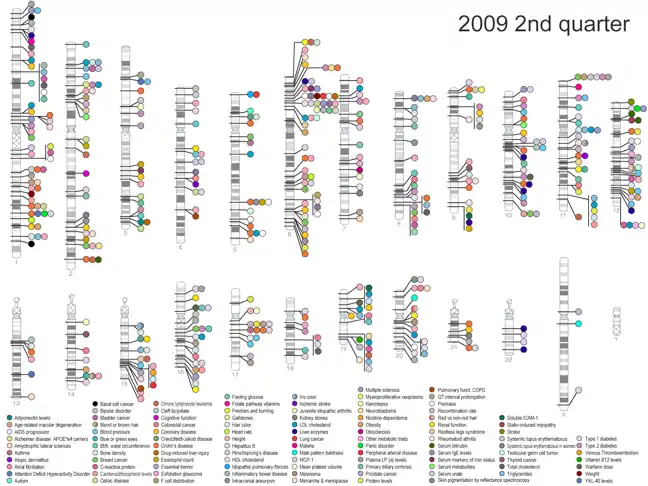
Tipos y variaciones
Mapeo de asociación en poblaciones donde se supone que los miembros son independientes
Varios métodos estándar para probar la asociación. Estudios de control de casos: los estudios de control de casos fueron uno de los primeros enfoques utilizados para determinar si una variante genética particular está asociada con un mayor riesgo de enfermedad en humanos. Al principio, Woofle en 1955, propuso una estadística de riesgo relativo que podría usarse para evaluar el riesgo dependiente del genotipo. Sin embargo, la preocupación persistente con respecto a estos estudios es la adecuación de los casos y controles coincidentes. En particular, la estratificación de la población puede producir asociaciones falsas positivas. En respuesta a esta preocupación, Falk y Rubenstein en 1987, sugirieron un método para evaluar el riesgo relativo que utiliza controles basados en la familia, obviando esta fuente de error potencial. Básicamente, el método utiliza una muestra de control de los alelos o haplotipos parentales no transmitidos a la descendencia afectada.
Mapeo de asociación en poblaciones donde se supone que los miembros están relacionados
En el mundo real es muy difícil encontrar individuos independientes (no relacionados). El mapeo de asociación basado en la población se ha modificado para controlar la estratificación de la población o la relación en el mapeo de asociación anidado. Todavía hay otra limitación en el mapeo QTL basado en la población; cuando la frecuencia del alelo favorable debe ser relativamente alta para ser detectada. Por lo general, los alelos favorables son alelos mutantes raros (por ejemplo, por lo general, un progenitor resistente podría ser 1 de cada 10000 genotipos). Otra variante del mapeo de asociación en poblaciones relacionadas es el mapeo de asociación basado en la familia. En el mapeo de asociación basado en la familia en lugar de múltiples individuos no relacionados, se utilizan múltiples familias o pedigríes no relacionadas. El mapeo de asociación basado en la familia[5] (enlace externo) se puede utilizar en situaciones en las que los alelos mutantes se han integrado en poblaciones. Uno de los populares mapas de asociación basados en la familia Prueba de desequilibrio de transmisión. Para más detalles, lea el mapeo QTL basado en la familia.
Ventajas
Las ventajas del mapeo de asociación basado en la población, utilizando una muestra de individuos de las colecciones de germoplasma o una población natural, sobre el mapeo QTL tradicional en cruces biparentales, se deben principalmente a la disponibilidad de variaciones genéticas más amplias con antecedentes más amplios para correlaciones de marcadores y rasgos. La ventaja del mapeo de asociación es que puede mapear rasgos cuantitativos con alta resolución de una manera estadísticamente muy poderosa. La resolución del mapeo depende de la extensión de LD, o asociación no aleatoria de marcadores, que ha ocurrido en todo el genoma. El mapeo de asociación ofrece la oportunidad de investigar material genético diverso e identificar potencialmente múltiples alelos y mecanismos de rasgos subyacentes. Utiliza eventos de recombinación que han ocurrido durante un período prolongado de tiempo. El mapeo de asociación permite la posibilidad de explotar datos de rasgos medidos históricamente para la asociación y, por último, no es necesario el desarrollo de poblaciones biparentales costosas y tediosas que hacen que el enfoque ahorre tiempo y sea rentable.[6][7]
Limitaciones
Un problema importante con los estudios de asociación es la tendencia a encontrar falsos positivos. Las poblaciones que muestran un rasgo deseado también llevan una variante genética específica no porque la variante realmente controle el rasgo, sino debido a la relación genética. En particular, las asociaciones indirectas que no son causales no se eliminarán al aumentar el tamaño de la muestra o el número de marcadores. Las principales fuentes de tales falsos positivos son el vínculo entre sitios causales y no causales, más de un sitio causal y epistasis. Estas asociaciones indirectas no se distribuyen aleatoriamente en todo el genoma y son menos comunes que los falsos positivos derivados de la estructura de la población.[8]
Asimismo, la estructura de la población siempre ha sido un problema constante. La estructura de la población conduce a asociaciones espurias entre los marcadores y el rasgo. Esto generalmente no es un problema en el análisis de enlaces porque los investigadores conocen la estructura genética de la familia que crearon. Pero en el mapeo de asociaciones, donde las relaciones entre poblaciones diversas no se entienden necesariamente bien, las asociaciones marcador-rasgo que surgen del parentesco y la historia evolutiva pueden confundirse fácilmente con las causales. Esto puede explicarse con modelos mixtos MLM. También llamado modelo Q + K, se desarrolló para reducir aún más la tasa de falsos positivos al controlar tanto la estructura de la población como la relación familiar críptica.[9]
Véase también
- Mapeo QTL
- Mapeo QTL basado en la familia
- Desequilibrio de ligamiento
- Estudio de asociación de genoma completo
- Asociación (ecología)
Referencias
- Gibson, G.; Muse S.V. (2009). A Primer of Genome Science. MA: Sinauer Associates.
- Hoeschele, I. (15 de julio de 2004). «Mapping Quantitative Trait Loci in Outbred Pedigrees». Handbook of Statistical Genetics. Chichester: John Wiley & Sons, Ltd. ISBN 978-0470022627. doi:10.1002/0470022620.bbc17.
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. (2008). «Genetic design and statistical power of nested association mapping in maize». Genetics 178 (1): 539-551. PMC 2206100. PMID 18202393. doi:10.1534/genetics.107.074245.
- Nussbaum, R.L.; McInnes, R.R.; Willard, H.F. (2007). Genetics in Medicine. Philadelphia, PA: Saunders Elsevier.
- Rosyara U.R., J.L. Gonzalez-Hernandez, K.D. Glover, K.R. Gedye and J.M. Stein. 2009.
- Abdurakhmonov I., Abdukarimov A. (2008).
- Kraakman, A. T. W. (1 de septiembre de 2004). «Linkage Disequilibrium Mapping of Yield and Yield Stability in Modern Spring Barley Cultivars». Genetics 168 (1): 435-446. ISSN 0016-6731. PMC 1448125. PMID 15454555. doi:10.1534/genetics.104.026831.
- Platt, A.; Vilhjalmsson, B. J.; Nordborg, M. (2 de septiembre de 2010). «Conditions Under Which Genome-Wide Association Studies Will be Positively Misleading». Genetics 186 (3): 1045-1052. ISSN 0016-6731. PMC 2975277. PMID 20813880. doi:10.1534/genetics.110.121665.
- Yu, Jianming; Pressoir, Gael; Briggs, William H; Vroh Bi, Irie; Yamasaki, Masanori; Doebley, John F; McMullen, Michael D; Gaut, Brandon S et al. (25 de diciembre de 2005). «A unified mixed-model method for association mapping that accounts for multiple levels of relatedness». Nature Genetics 38 (2): 203-208. ISSN 1061-4036. PMID 16380716. doi:10.1038/ng1702.
| Control de autoridades |
|
|---|
 Datos: Q4809584
Datos: Q4809584