Paraganglioma carotídeo
El paraganglioma es un tumor que derivana de las células cromafines. Se localizan en la adventicia de las estructuras vasculares y neuronales, generalmente en las proximidades de los ganglios del sistema nervioso autónomo, esta característica es la que les da su nombre. Tiene una incidencia de entre 1/30.000 y 1/100.000.
| Paraganglioma carotídeo | ||
|---|---|---|
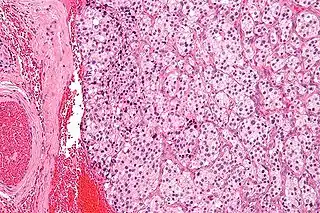 Micrografía de un tumor de cuerpo carotídeo (un tipo de paraganglioma). | ||
| Especialidad | oncología | |
Los paragangliomas que derivan de la médula adrenal son los más frecuente y reciben un nombre diferente, feocromocitomas. El resto son paragangliomas extraadrenales.[1][2] La terminología recomendada por la Organización Mundial de la Salud (OMS) para denominar los tumores de los paraganglios extraadrenales es la de “paraganglioma” seguido de la localización anatómica en la que se encuentra, indicando si además el tumor es funcionante o maligno. La clasificación de la OMS consta de 13 tipos: del cuerpo carotídeo, yugulo-timpánico, vagal, laríngeo, aórtico-pulmonar, gangliocítico, de la cauda equina, orbital, nasofaríngeo, extraadrenal simpático, paraaórtico, vesical y paravertebral (intratorácico y cervical). Según esta clasificación los paragangliomas carotídeos, también denominados tumores del cuerpo carotídeo, están incluidos en el grupo de paragangliomas de cabeza y cuello.[3]
Los paragangliomas carotídeos son muy vascularizados, de crecimiento lento e invasivos hacia las estructuras próximas. Los paraganglios de los que proceden se encuentran localizados en la adventicia de la bifurcación de la arteria carótida común (CCA) constituyendo un órgano altamente especializado, que se nutre por ramos arteriales procedentes de la carótida externa. El cuerpo carotídeo actúa como un órgano quimiorreceptor vascular participando en el control autónomo de los sistemas respiratorio y cardiovascular y en el control de la temperatura de la sangre.[4]
Clasificación de Shamblin
Shamblin et al. propusieron en 1971 una clasificación de los CBTs basada en el tamaño del tumor y la invasión de la arteria carótida, lo que permite evaluar las probabilidades de resección. Según estos criterios dividieron los tumores en tres grupos distintos:
- Grupo 1: se encuentran bien localizados, no invaden los vasos mayores adyacentes y son tumores pequeños, fácilmente disecables.
- Grupo 2: adheridos o rodeando parcialmente los vasos. Comprimen la arteria carótida interna y arteria carótida externa pero pueden ser disecable de los vasos mediante cuidadosa disección subadventicial.
- Grupo 3: presentan un gran tamaño, involucran ampliamente las carótidas y estructuras adyacentes. Para su exéresis completa se requiere resección parcial o total de estas arterias.[5]
Revisión de artículos sobre la etiología del paraganglioma carotídeo
Para determinar la etiología de los PG, es preciso establecer dos grandes grupos en este tipo de neoplasias. La revisión de Soto et al distingue PG carotídeos esporádicos y familiares.[6] Según Lack et al, los PG familiares constituyen el 10% de los casos y entre un 35 y un 50% de éstos se asocian con multicentricidad, lo que determina la importancia de un cuidadoso estudio de estos pacientes.[7] Para Lee et al, el porcentaje de casos de PG familiares con multicentricidad se sitúa en el 10%.[8] Según la revisión referida por Soto et al, los tumores familiares suponen entre el 10 y el 35% de los casos, son bilaterales y están asociados con el síndrome de neoplasia endocrina múltiple tipo II (MEN II) con herencia ligada al sexo masculino. Este artículo también describe una mayor incidencia de PG esporádicos, y lo apoya la misma información reportada en el artículo de Devender Singh et al.,[6][9] Para Antúnez et al, los casos esporádicos tienen mayor predisposición a la malignidad que los familiares, señalando también que las metástasis pueden aparecer hasta en los 20 años sucesivos a la lesión primaria.[10] Sin embargo, Sánchez de Guzmán et al atribuye una menor malignidad a los casos esporádicos, relacionándolos con una menor frecuencia de PG secretores y de bilateralidad.[11] Soto et al, apoya este último dato al reportar que su único caso bilateral, de los 10 que componen su estudio, se incluía en el contexto de PG familiar. Este mismo artículo refiere asociación entre PG esporádicos y unilateralidad.[6] Apoya esta vertiente el artículo de Sevilla García et al, que relaciona de nuevo los PG familiares con un mayor potencial maligno, en especial los tumores situados en la vecindad del órgano de Zuckerkandl, que suponen un 1,14% de todos los PC.[12]
Aunque los PG son una entidad genéticamente heterogénea, hasta el momento se han identificado 4 locus cromosómicos relacionados con la enfermedad, que según la revisión de Sevilla García et al son: 11q23 (PGL1), 11q13(PGL2), 1q21 (PGL3) y 1p36.1p35 (PGL4).[13] Gracias a estudios de asociación, se han determinado los genes correspondientes a 3 de estos locus: SDHD en 11q23, SDHC en 1q21 y SDHB en 1p36.1p35; sin haberse esclarecido el gen del locus PGL2. Lozano Sánchez et al también describe la implicación etiopatogénica de los genes codificantes para tres de las cuatro subunidades de la enzima succinato deshidrogenasa (SDH), dentro del complejo mitocondrial II, en el desarrollo de PG y otras entidades asociadas.[14] Las revisiones de Sato et al y de Wieneke et al, informan con estos mismos datos.[15][16] Sevilla García et al, menciona su papel crucial tanto en el ciclo de Krebs como en la fosforilación oxidativa, y explica cómo la inactivación por mutación sobre SDH produce un efecto similar a la estimulación por hipoxia crónica en las células paraganglionares. Las mutaciones SDHD y SDHB incrementan la concentración intracelular de mediadores moleculares de hipoxia (HIF) y estimulan genes que promueven la angiogénesis (VEGF), lo que se traduce en una mayor proliferación celular hiperplásica que desencadena la neoplasia.[13] También el artículo de Sato et al señala el mal funcionamiento mitocondrial, la situación de hipoxia y la angiogénesis como causa directa de la aparición del tumor en relación con dicha mutación.[15] De acuerdo con la información citada por Kollert el al, estas alteraciones siguen un patrón de herencia autosómica dominante.[17] Este dato también se menciona en el artículo de Lack et al, quien además añade el de la penetrancia incompleta en su transmisión a la descendencia.[7] Un amplio estudio de casos europeo y americano llevado a cabo por Neumann et al, incluyó 83 PG de cabeza y cuello, y concluyó que en la mutación SDHD son más incidentes los PG multifocales, incluidos los silentes; mientras que la mutación para SDHB se asocia con mayor malignidad.[18] Para Astuti et al, esta última mutación se relacionan con susceptibilidad al desarrollo de PG intraadrenales, extraadrenales y de cabeza y cuello.[19] Mientras Benn et al los asocia con una localización extraadrenal y fuerte tendencia a la metástasis.[20] Los datos del artículo de Giménez-Roqueplo et al siguen la misma línea: de 84 pacientes con PG esporádicos, 8 presentaron esta mutación, y 7 de estos 8 se relacionaron con desarrollo tumoral extraadrenal, con malignidad, y con recurrencia.[21] 8 de los 29 pacientes seguidos por Timmers et al con PG relacionados con esta mutación desarrollaron metástasis entre los 2,7 y 4,1 años posteriores al diagnóstico.[22] Sato et al describe un caso de mutación SDHB con wildtype para SDHD en un paciente con PG extraadrenal no metastásico.[15] Neumann et al publican como resultados de un estudio de cohortes sobre 34 pacientes con SDHD un 74% de PG multifocales y benignos. Este mismo artículo reporta altas tasas de malignidad relacionadas con la mutación SDHB, con un 34% de sus casos.[18] Este porcentaje llega al 71% en el estudio de Amar et al siendo menor, 37,5%, el reportado por Benn et al.[23][20] Con todo esto, parece muy razonable la sentencia de Sato et al, que recomienda el seguimiento postoperatorio indefinido en pacientes con resección de un PG asociado a mutación SDHB.[15] Sobre la mutación SDHC, los artículos de Manelli et al y de Erlic et al, sostienen una relación más importante con PG de cabeza y cuello, y otra más infrecuente con PG intraadrenal.,[24][25] Sato et al, señala la menor incidencia de este tipo de alteración.[15] Aunque la mutación sobre la subunidad A de la SDH, no se ha relacionado con PG ni otras neoplasias, sí lo ha hecho con una encefalopatía juvenil autosómica recesiva, citando el artículo de Bourgeron et al.[26]
Notas y referencias
- Antúnez, A. (2009). «Paragangliomas cervicocefálicos: Anatomía Patológica de los paragangliomas cervicocefálicos». Revista acta otorringolaringológica española 60.
- Giovanni, A. (2007). «Cervical paragangliomas: is SDH genetic analysis systematically required?. Eur Arch Otorhinolaryngol». Eur Arch Otorhinolaryngol. 265.
- Ramírez, JL. (2009). «Paragangliomas: Métodos de imagen y correlación histopatológica». Anales de Radiología México 4.
- Ortega, C. (2006). «Diagnóstico y tratamiento de los paragangliomas carotídeos. Presentación de nueve casos y revisión de la literatura». Acta Otorrinolaringología Española (57).
- Espinel-Ortiz, C (2008). «Paragangliomas de cuerpo carotídeo: clasificación y manejo de 143 tumores.». Acta Otorrinolaringología & Cirugía de Cabeza y cuello 3 (36).
- Soto, S. (2007). Tumores del cuerpo carotídeo: a propósito de 10 casos tratados.
- Lack, E. (1997). Tumors of the adrenal gland and extra-adrenal paraganglia.
- Lee, K.Y. (2006). Extraadrenal paragangliomas of the body: imaging features.
- Singh, D. (2006). Management for carotid body paragangliomas. Interactive CardioVascular and Thoracic Surgery.
- Antúnez, A. (2009). Paragangliomas cervicocefálicos: Anatomía Patológica de los paragangliomas cervicocefálicos.
- Sánchez De Guzmán, G. (2008). Paragangliomas de cuerpo carotídeo: clasificación y manejo de 143 tumores.
- Singh, D. (2006). Management for carotid body paragangliomas. Interactive CardioVascular and Thoracic Surgery.
- Sevilla García, M.A. (2007) Paragangliomas de cabeza y cuello: revisión de 89 casos en 73 pacientes.
- Lozano-Sánchez, F.S. (2009) Tratamiento quirúrgico de los paragangliomas carotídeos.
- Sato, H. (2010). L157X nonsense mutation of the succinate dehydrogenase subunit B gene in a Japanese patient with right paraaortic paraganglioma.
- Wieneke, J.A. (2009) Paraganglioma: Carotid Body Tumor.
- Kollert, M. (2006). Cervical Paragangliomas. Tumor Control and Long-Term Functional Results after Surgery.
- Neumann, H.P. (2004) Distinct clinical features of paragangliomas syndrome associated with SDHB and SDHD gene mutations.
- Astuti, D. (2001). Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma.
- Benn, D.E. (2006). Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes.
- Gimenez-Roqueplo, A.P. (2003). Mutations in the SDHB gene are associated with extra-adrenal and/or malignant pheochromocytomas.
- Timmers, H.J.L. (2007). Clinical presentations, biochemical phenotypes, and genotype-phenotype correlation in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas.
- Amar, L. (2005). Genetic testing in pheochromocytoma or functional paraganglioma.
- Mannelli, M. (2007). Genetic screening for pheochromocytoma: should SDHC gene analysis be included?
- Erlic, Z. (2009). Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients.
- Bourgeron, T. (1995). Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency.
| Control de autoridades |
|
|---|
 Datos: Q581592
Datos: Q581592 Multimedia: Paraganglioma / Q581592
Multimedia: Paraganglioma / Q581592