Proteinosis alveolar pulmonar
La proteinosis alveolar pulmonar (PAP) o fosfolipoproteinosis alveolar pulmonar es la acumulación anormal de material eosinofílico rico en lípidos procedente del surfactante pulmonar en los alvéolos, interfiriendo en la hematosis. Puede ocurrir en forma primaria o secundariamente en el marco de malignidad (especialmente en leucemia mieloide), infección pulmonar o exposición ambiental a polvos o químicos. Se han reconocido raras formas familiares, lo que sugiere un compontente genético en ciertos casos. El padecimiento es más común en hombres que en mujeres y particularmente entre los 30 y 50 años, aunque se puede presentar desde la infancia hasta la tercera edad.[1]
| Proteinosis alveolar pulmonar | ||
|---|---|---|
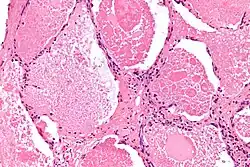 Micrografía de una proteinosis alveolar pulmonar con tinción hematoxilina-eosina. | ||
| Especialidad | Neumología | |
Manifestaciones clínicas
El inicio de la enfermedad es insidioso y crónico. En promedio, la evolución sintomática previo al diagnóstico es de siete meses. Entre los síntomas más comunes son la disnea y tos (regularmente seca y, en raras ocasiones, con hemoptisis). En menor medida, se presenta dolor torácico, fiebre y pérdida de peso. En el examen físico, no se presentan signos anormales. Aunque, en algunos casos se pueden escuchar estertores crepitantes en la exploración de tórax. En alrededor del 25% de los casos, se presenta acropaquia y cianosis.[1]
Independientemente de las manifestaciones clínicas, la PAP se caracteriza por la acumulación de material lipoproteináceo procedente del surfactante en los alvéolos pulmonares[2] con preservación del intersticio pulmonar.[3]
Mecanismo
Se cree que la PAP causada por una combinación de factores genéticos y adquiridos que perjudican el metabolismo del surfactante pulmonar.[1] La patogenia está relacionada con alteraciones en la homeostasis de esta sustancia, de la que hay una producción normal, pero existen defectos en su aclaramiento por parte de los macrófagos alveolares.[4]
Existen al menos tres mecanismos fisiopatológicos del PAP: excesiva acumulación de lipoproteínas del surfactante en los alvéolos, casos congénitos presentados en el periodo neonatal con hipoxemia grave y defectos genéticos en la proteína del surfactante o en la molécula del receptor βc del factor estimulante de colonias de granulocitos y macrófagos (GM-CSF).[1] El surfactante disfuncional se genera a partir de mutaciones en los genes codificantes de las proteínas B o C del surfactante o a causa de mutaciones de la cadena βc del GM-CSF.[2]
Diagnóstico
En la radiografía se muestran patrones radiográficos no específicos de consolidación bilateral y dispersa del parénquima pulmonar. No se muestran signos específicos que sugieran el PAP.[5] Por su parte, en la tomografía se observa un patrón característico conocido como «pavientación» con distribución «en parches o geométrica». El diagnóstico requiere una biopsia pulmonar por cirugía o endoscopia. Ante un examen microscópico, estas muestran presentan «espacios alveolares llenos con un material eosinofílico granular» y positivo a la técnica de Schiff. Igualmente, los exámenes de muestras alveolares obtenidas por lavado broncoalveolar exhiben «tinción granular eosinofílica difusa» con pocos macrófagos y positivo a la técnica PAS.[1] La prueba ELISA para anticuerpos contra el GM-CSF puede emplearse para el diagnóstico de PAP autoinmune.[6]
Tratamiento
Aunque el 25% de los casos pueden resultar en remisión espontánea, se puede iniciar un tratamiento si el paciente sufre limitaciones por la disnea. El tratamiento estándar para la PAP es el lavado broncoalveolar con solución salina. En pacientes adultos se pueden requerir grandes volúmenes y repetir el procedimiento.[2][3] Es además el tratamiento más seguro y efectivo.[5]
Referencias
- Mejía A., Mayra H.; Alonso M., Delfino; Suárez L., Teresa de J.; Estrada G., Andrea; Gaxiola G., Miguel O.; Carrillo R., J. Guillermo (2006). «Proteinosis alveolar pulmonar». Neumología y Cirugía de Tórax 65 (S3): S15—S23. Consultado el 8 de marzo de 2016.
- Param, Teresa; Flores, Manuel; Bolbaran, Juan Pablo; Díaz, Armando; Acuña, Juan Carlos; Ochoa, Laura (2007). «Proteinosis alveolar pulmonar, caso clínico». Revista Chilena de Pediatría 78 (2): 176-182. doi:10.4067/S0370-41062007000200009.
- Barraclough, R. M.; Gillies, A. J. (2001). «Pulmonary alveolar proteinosis: a complete response to GM-CSF therapy». Thorax 56 (8): 664-665. PMC 1746117. PMID 11462071. doi:10.1136/thorax.56.8.664. Consultado el 8 de marzo de 2016.
- Rodríguez Portal, José Antonio; Rodríguez Becerra, Eulogio; Sánchez Garrido, Antonio (2009). «Proteinosis alveolar. Respuesta al tratamiento con factor estimulante de colonias de granulocitos y macrófagos por vía inhalada». Archivos de Bronconeumología 45 (3): 150-152. PMID 19286115. doi:10.1016/j.arbres.2008.02.006.
- Shah, Pallav L.; Hansell, David; Lawson, Peter R.; Reid, Kenneth B. M.; Morgan, Cliff (2000). «Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis». Thorax 55 (1): 67-77. PMC 1745595. PMID 10607805. doi:10.1136/thorax.55.1.67. Consultado el 8 de marzo de 2016.
- Uchida, Kanji et al. (2014). «Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis». Journal of Immunological Methods 402 (1-2): 57-70. PMID 24275678. doi:10.1016/j.jim.2013.11.011.
| Control de autoridades |
|
|---|
 Datos: Q448698
Datos: Q448698 Multimedia: Pulmonary alveolar proteinosis / Q448698
Multimedia: Pulmonary alveolar proteinosis / Q448698