Reacción de Alder-eno
La reacción Alder-eno (llamada también Reacción eno) es una reacción química orgánica entre un alqueno que presente un hidrógeno alílico y un compuesto con al menos un enlace π llamado enófilo. La reacción consiste en la migración 1,5 del hidrógeno alílico con la formación de un enlace σ. El producto obtenido es un alqueno con el doble enlace intercambiado en la posición alílica. El enófilo puede ser un alqueno, un aldehído, una cetona o una imina.[1]
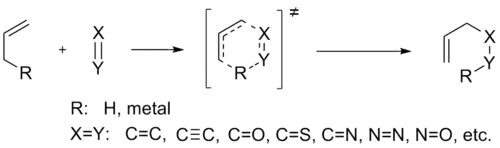
La reacción Alder-eno presenta un mecanismo relacionado con el de la reacción de Diels-Alder, pero las energías de activación son mucho mayores. Como resultado, las reacciones eno por lo general requieren altas temperaturas, lo que ha limitado la propuesta de los mecanismos y su uso en síntesis.[2] A temperatura mayores a 400 °C se lleva a cabo la reacción inversa, la reacción Retro-Eno. Sin embargo, muchas reacciones catalizadas por ácidos de Lewis han resultado útiles y se han desarrollado métodos que pueden tener altos rendimientos y selectividades a temperaturas significativamente más bajas. En general, el proceso eno se ve favorecida por sustituyentes electroatractores en el enófilo, por la tensión generada por el componente eno. La reacción procede cuando los dos reactivos se alinean en una geometría favorable.[3]
El componente eno
El componente eno puede ser una molécula que contenga un enlace π y un hidrógeno activo en la posición alílica, propargílica o α. Los posibles grupos funcionales que pueden funcionar como componentes eno incluyen a los olefínicos, acetilénicos, alénicos, aromáticos, ciclopropilos, y hetero-carbono. [4] Por lo general, el hidrógeno alílico de los componentes alénicos participan en las reacciones eno, pero en el caso de los alenilsilanos, el átomo de hidrógeno alénico α al sustituyente de silicio es el transferido, dándose así un sililalquino. El fenol puede actuar como un componente eno, por ejemplo en la reacción con dihidropirano, pero se requieren altas temperaturas (150-170 °C). Sin embargo, los grupos eno con alta tensión y anillos pequeños fusionados sufren reacciones eno a temperaturas mucho más bajas. Además, los componentes que contienen los enlaces carbonilo (C = O), imino (C = N) y tiocarbonilo (C = S) han sido reportados, pero tales casos son raros.[4]
El enófilo
Los enófilos son moléculas que contienen al menos un enlace π con sustituyentes electroatractores que reducen significativamente el Orbital más Bajo Desocupado (miniatura) del enlace π. Los enófilos pueden contener enlaces carbono-carbono múltiples (olefinas, acetilenos, benzinos), enlaces carbono-hetero múltiples (C = O en el caso de las reacciones carbonil-eno, C = N, C = S, C ≡ P), enlaces múltiples hetero-hetero (N = N, O = O, Si = Sí, N = O, S = O), sistemas de cumuleno (N = S = O, N = S = N, C = C = O, C = C = S, SO2) y sistemas π con carga formal positiva (C=N+, C=S+, C≡O+, C≡N+).[4]
Mecanismo
La reacción Alder-eno es una reacción pericíclica. El modo de adición del enófilo al grupo eno en la reacción Alder-eno térmica puede ser descrito como suprafacial, donde se da la interacción de tres componentes: los orbitales HOMO del eno, el orbital LUMO del enlace CH alílico del ene y el LUMO del enófilo (Figura 2).[5]
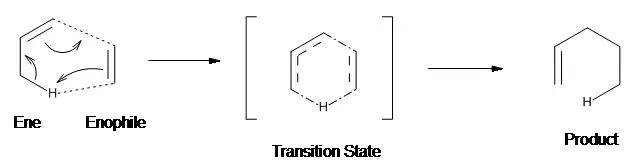
La naturaleza concertada del proceso eno ha sido sustentada experimentalmente,[6] y la reacción puede ser designada como [σ2s + π2s + π2s] en la notación de Woodward-Hoffmann.[5] El estado de transición propuesto para la reacción eno térmica del propeno con el formaldehído tiene una conformación de sobre, con un ángulo de enlace C-O-H de 155ºo, tal y como fue calculado en el nivel 3-21G.[7]
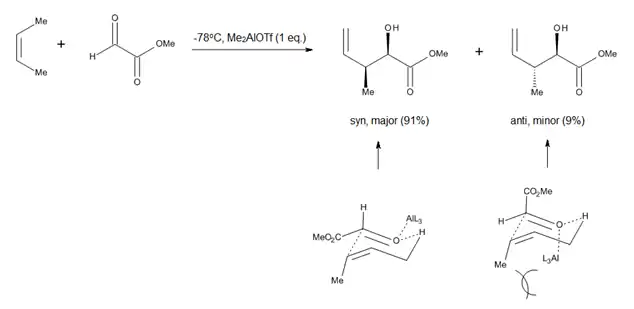
El estudio de las reacciones de carbonilos promovidos por ácidos de Lewis, tales como los procesos glioxilato-eno catalizados por aluminio, (Figura 4), ha alentado a considerar una conformación parecida al de silla para el estado de transición. La ventaja de este modelo es el hecho de que los parámetros estéricos tales como 1,3-diaxial y la repulsión 1,2-diecuatorial son fáciles de visualizar, lo que permite predicciones precisas sobre la diastereoselectividad de muchas reacciones.[3]
Mecanismo birradical
Cuando un mecanismo concertado es geométricamente desfavorable, una reacción térmica eno puede ocurrir a través de una vía birradical no concertada, sino por pasos. Por ejemplo, la reacción eno del ciclopenteno y ciclohexeno con azodicarboxilato de dietilo puede ser catalizada por iniciadores de radicales libres. El carácter gradual del proceso se ve favorecido por la estabilidad de los radicales ciclopentenilo y ciclohexenilo, así como la dificultad de ciclopenteno y ciclohexeno de obtener la geometría óptima de un proceso concertado.[8]
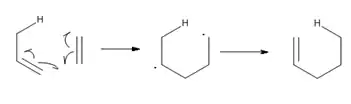
Regioselectividad
Así como en el caso de cicloadición, el éxito de una reacción eno es determinada en gran medida por la accesibilidad estérica del hidrógeno alílico del eno. En general, los átomos de hidrógenos metílicos y metilénicos se abstraen mucho más fácilmente que el hidrógeno del metino. En las reacciones térmicas eno, el orden de reactividad de la abstracción del átomo de Hidrógeno es primario> secundario> terciario, independientemente de la estabilidad termodinámica de los productos de olefinas internas. En reacciones promovidas por ácidos de Lewis, el par enófilo - ácido de Lewis determina en gran medida la relativa facilidad de abstracción de hidrógenos de metilo vs metileno.[3]
La orientación de una adición eno puede predecirse a partir de la estabilización relativa de las cargas parciales desarrolladas en un estado de transición asimétrica, con la consecuente formación del enlace σ. El regioisómero más favorecido vendrá del estado de transición en el que las cargas transitorios son más estables por la orientación del par eno-enófilo.[4]
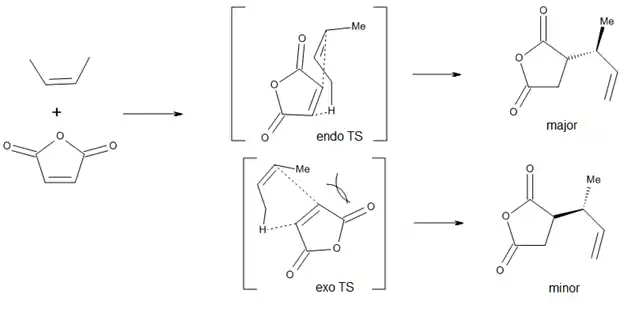
Inducción asimétrica interna
Respecto a la diastereoselectividad con respecto a los nuevos centros quirales formados, se ha observado una preferencia cualitativa al isómero endo, pero los efectos estéricos pueden modificar esta preferencia (Figura 6).[3]
Reacciones eno intramoleculares
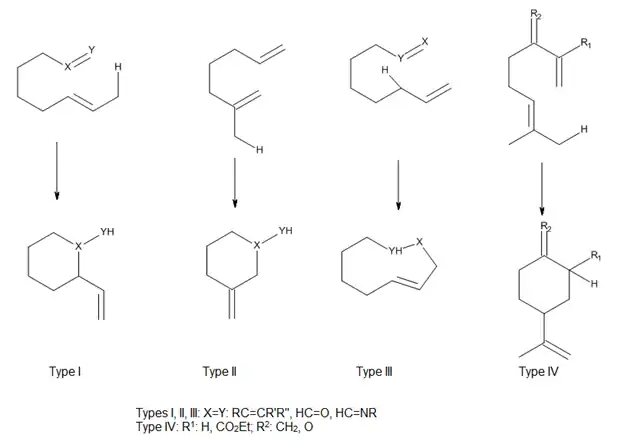
Las reacciones eno intramoleculares proceden debido a que tienen entropías de activación negativas menores que sus contrapartes intrermoleculares, por lo que proceden con mayor facilidad; ocurren incluso en los casos de enófilos simples, tales como alquenos no activados y alquinos.[9] Las altas regio-y estereoselectividades que se pueden obtener en estas reacciones pueden ofrecer un control considerable en la síntesis de sistemas de anillos complicados.
Teniendo en cuenta la posición de conexión del ene y el enófilo, Oppolzer ha clasificado a las reacciones eno térmicas y catalizadas por ácidos de Lewis intramoleculares como los tipos I, II y III. Snider ha añadido un tipo de reacción IV (Figura 7). En estas reacciones, el solapamiento orbital entre la eno y enológico es en gran parte controlado por la geometría de la aproximación de los componentes.[4]
Reacciones eno catalizadas por ácidos de Lewis
Las reacciones eno térmicas tienen varios inconvenientes, tales como la necesidad de temperaturas muy altas y la posibilidad de reacciones secundarias, como la polimerización de olefinas catalizada por protones o reacciones de isomerización. Ya que los enófilos son deficientes en electrones, se ha inferido que la formación de complejos con los ácidos de Lewis debe acelerar la reacción eno, como ocurre en la reacción que se muestra en la Figura 8.
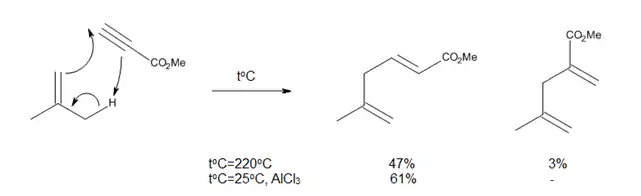
Los halogenuros de alilaluminio son bien conocidos como aceptores de protones, y su uso como catalizadores ácidos de Lewis en las reacciones eno ha ampliado el alcance de estas reacciones y ha permitido su estudio y el desarrollo en condiciones mucho más suaves.[2]
Ya que un ácido de Lewis puede acomplejarse directamente con el oxígeno del carbonilo, los catalizadores de trialquilaluminio se han desarrollado para enófilos que contienen un enlace C = O. En particular, se encontró que Me2AlCl es un catalizador muy útil para las reacciones eno para aldehídos y cetonas α, β-insaturados, además de otros aldehídos alifáticos y aromáticos. La razón del éxito de este catalizador es el hecho de que el complejo eno-aducto-Me2AlCl pueden reaccionar para producir el metano y el alcóxido de aluminio, los cual previene la transposición catalizada por ácidos y la solvólisis (Figura 9).[2]
Condiciones de reacción
Siempre y cuando la nucleofilicidad del grupo alquilo no lleve a reacciones secundarias, son suficientes cantidades catalíticas de ácido de Lewis para muchas reacciones eno con enófilos reactivos. Sin embargo, la cantidad del ácido de Lewis puede variar ampliamente, ya que depende en gran medida la basicidad relativa de los enófilos y el aducto eno. En cuanto a la elección de disolvente para las reacciones, los rendimientos más altos se alcanzan habitualmente a utilizar halocarbonos como disolventes. Los solventes polares, tales como los éteres, no son adecuados ya que pueden formar complejos con el ácido de Lewis, lo que inactiva al catalizador).[2]
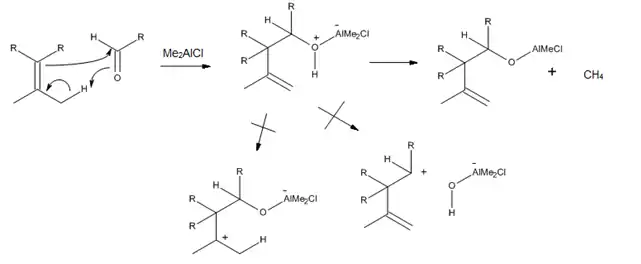
Reactividad de los grupos eno
Mientras que los efectos estéricos siguen siendo importantes para determinar el resultado de una reacción eno catalizada por ácidos de Lewis, los efectos electrónicos son también importantes, ya que en la reacción habrá una carga positiva considerable desarrollada en el carbono central del eno. Como resultado, los alquenos con al menos un carbono vinílico disustituidos son mucho más reactivos que los mono o los 1,2 disustituidos.
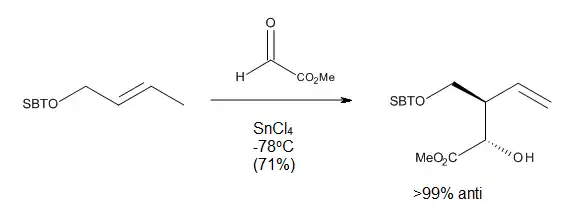
Mecanismo
Como se observa en la Figura 11, las reacciones eno catalizadas por ácidos de Lewis pueden proceder ya sea a través de un mecanismo concertado que presenta un estado de transición polar, o a través de un mecanismo paso a paso con un intermediario zwitteriónico. El eno, enófilo y el catalizador elegido pueden influir en que la vía es el proceso energético más bajo. En general, el complejo del grupo eno o el enófilo con el ácido de Lewis es más reactivo, aunque también es probable que la reacción sea gradual.[2]
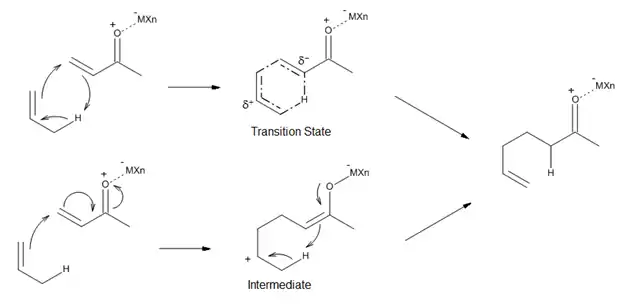
Catálisis asimétrica
Una dirección actual en el estudio de las reacciones eno catalizadas por ácidos de Lewis es el desarrollo de catalizadores asimétricos para la formación de enlaces C-C. Un ejemplos es un complejo de titanio quiral (R)-1a, que fue desarrollado por Mikami[10] para las reacciones eno asimétricas proquirales con participación de glioxilato. El catalizador se prepara in situ a partir de i-PrO)2TiX2 y binaftol ópticamente puro; el intercambio alcoxi-ligando es facilitado por el uso de tamices moleculares. El método permite obtener α- hidroxiésteres de alta pureza enantiomérica, los cuales representan una clase de compuestos de importancia biológica y sintética (Figura 12).[10]
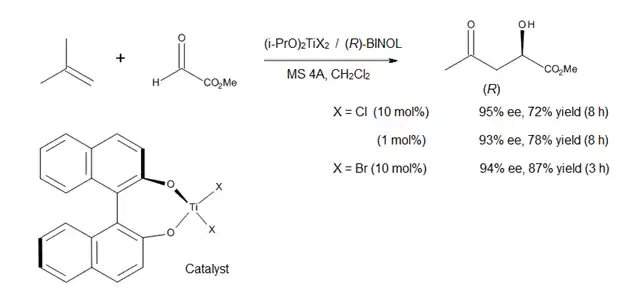
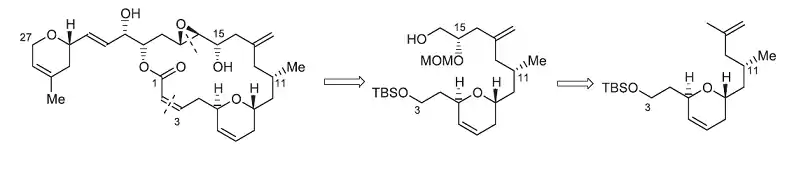
La síntesis total del laulimálido[11] (compuesto 1, Figura 13) ilustra cuán útil puede resultar el método de Mikami. El laulimálido es un metabolito de diversas esponjas que podrían tener un uso potencial como un agente antitumoral, debido a su capacidad para estabilizar los microtúbulos. Uno de los pasos clave en la estrategia utilizada para la síntesis del fragmento de C3-C16 fue una reacción eno catalizada quiralmente que instaló el estereocentro C15. El tratamiento del grupo terminal alilo del compuesto 3 con glioxilato de etilo en presencia de catalizadores (S)-BINOL-TiBr2 produjo el alcohol requerido con rendimiento de 74% y una ds> 95%. Este método elimina la necesidad de un grupo protector o cualquier otra funcionalidad en el extremo de la molécula. Además, al llevar a cabo esta reacción, Pitts et al. logró evitar las duras condiciones y bajos rendimientos asociados a la instalación de unidades exo-metileno a finales de la síntesis.
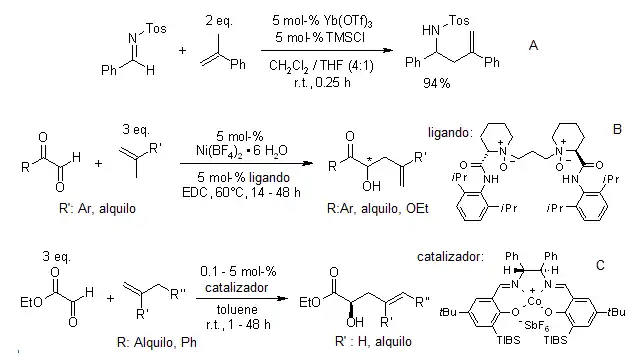
Otros métodos han sido reportados, como el uso de complejos de iterbio (Yamanaka),[12] cobalto (Hutson)[13] y níquel (Zheng)[14] como ácidos de Lewis.
Véase también
Referencias
- Alder, K.; Pascher, F; Schmitz, A. (1943). Ber. dtsch. chem. Ges 76: 27.
- Snider, B. B. (1980). Acc. Chem. Res. 13: 426.
- Mikami, K.; Shimizu, M. (1992). Chem. Rev. 92: 1021.
- Paderes, G. D.; Jorgensen, W. L. (1992). J. Org. Chem. 57: 1904 and references within.
- Inagaki, S.; Fujimoto, H; Fukui, K. J. (1976). J. Am. Chem. Soc. 41: 4693.
- Stephenson, L. M.; Mattern, D. L. (1976). J. Org. Chem. 41: 3614.
- Loncharich, R. J.; Houk, K. N. (1987). J. Am. Chem. Soc. 109: 6947.
- Thaler, W. A.; Franzus, B. J. (1964). J. Org. Chem. 29: 2226.
- Oppolzer, W.; Snieckus, V. (1978). Angew. Chem. Int. Ed. Engl. 17: 476.
- Mikami, K.; Terada, M.; Takeshi, N. (1990). J. Am. Chem. Soc. 112: 3949.
- Pitts, M. R.; Mulzer, J. (2002). Tetrahedron Letters 43: 8471.
- Yamakana, M.; A. Nishida; M. Nakagawa (2000). Organic Letters 2: 159-161.
- Hutson, G.E.; A. H. Dave, V. H. Rawal (2007). Organic Letters 9: 3869.
- Zheng, K.; J. Shi; X. Liu; X. Feng (2008). J. Am. Chem. Soc. 130: 15770.
| Control de autoridades |
|
|---|
 Datos: Q26469
Datos: Q26469 Multimedia: Ene reactions / Q26469
Multimedia: Ene reactions / Q26469