Síndrome de DiGeorge
El síndrome de deleción 22q11.2, también llamado síndrome de DiGeorge,[1][2][3] es una enfermedad causada por la deleción de una pequeña parte del cromosoma 22. Esta deleción se presenta cerca a la mitad del cromosoma en la ubicación designada como q11.2, en uno de los brazos largos de cualquiera de los dos cromosomas 22. Tiene una prevalencia estimada de 1:4000.[4] El síndrome fue descrito en 1968 por el pediatra endocrinólogo estadounidense Angelo DiGeorge.[5][6] Amplificación de sonda dependiente de la ligadura multiplex
| Síndrome de DiGeorge | ||
|---|---|---|
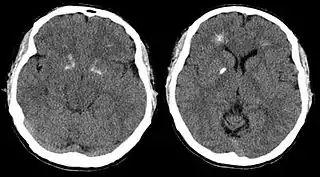 Corte por tomografía axial computarizada de un paciente que muestra calcificaciones periventriculares y de los ganglios basales | ||
| Especialidad | genética médica | |
| Sinónimos | ||
|
Síndrome velocardiofacial Síndrome de Shprintzen Síndrome de anomalía facial conotruncal Síndrome de Strong | ||
El síndrome de deleción 22q11.2 tiene una herencia autosómica dominante. Alrededor del 7 % de todos los casos de este síndrome son heredados del padre, teniendo sus descendientes una probabilidad del 50 % de heredarla.
El síndrome de DiGeorge presenta una amplia variedad de síntomas, aunque no todos están presentes en un determinado paciente. Por lo general existen infecciones recurrentes, defectos cardíacos, hipocalcemia, una constitución facial característica y problemas de paladar o insuficiencia velofaríngea. Raramente se presenta en los individuos sarcoma de Kaposi. Estos síntomas se producen debido a una maduración muy pobre o nula de linfocitos T por parte del timo.
Las alteraciones en el cromosoma 22, pueden estar ligadas a su vez a mayor susceptibilidad para la esquizofrenia, así como, se asocia con la ataxia 10 espinocerebelosa (SCA10).
Diagnóstico molecular

Su diagnóstico molecular consiste en la realización de varios tipos de pruebas genéticas:
- Hibridación fluorescente in situ (FISH): Normalmente se utilizan dos sondas que se encuentran comercialmente disponibles para el análisis, TUPLE1 y N25. El único inconveniente de estas sondas es que no son lo suficientemente sensibles como para detectar deleciones pequeñas (<40 kb) en 22q11.2. En algunas condiciones también se puede utilizar la sonda ARSA, siempre y cuando sean en casos de control.
- Amplificación de sonda dependiente de la ligadura multiplex (MLPA): Utilizando el kit P250 es posible detectar la deleción típica y deleciones pequeñas. Es mucho más específica que el FISH, y también es el método de confirmación de las deleciones detectadas por array CGH.
- Otros métodos de diagnóstico también incluyen técnicas como array CGH y PCR cuantitativa.
Referencias
- Orphanet: Monosomy 22q11. Consultado el 15 de junio de 2012
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- Oskarsdóttir S, Vujic M, Fasth A (2004). «Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden». Arch. Dis. Child. 89 (2): 148-51. PMC 1719787. PMID 14736631. doi:10.1136/adc.2003.026880.
- DiGeorge AM. Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism. IV(1). White Plains, NY: March of Dimes-Birth Defects Foundation; 1968:116-21
- Restivo, Angelo; Sarkozy, Anna; Digilio, Maria Cristina; Dallapiccola, Bruno; Marino, Bruno (febrero de 2006). «22q11 Deletion syndrome: a review of some developmental biology aspects of the cardiovascular system.». Journal of Cardiovascular Medicine 7 (2): 77-85. PMID 16645366. doi:10.2459/01.JCM.0000203848.90267.3e.
Enlaces externos
| Control de autoridades |
|
|---|
 Datos: Q525642
Datos: Q525642 Multimedia: DiGeorge syndrome / Q525642
Multimedia: DiGeorge syndrome / Q525642