Síndrome de Joubert
El síndrome de Joubert es una rara anomalía de carácter genético, mayoritariamente autosómica recesiva que afecta al cerebelo y puede ser confundida con otras afecciones, como el autismo. Aparece por la ausencia o el bajo grado de desarrollo del vermis del cerebelo (en este caso, los cilios de la membrana plasmática de sus células, que mueven el líquido cefálico) debido a factores de carácter genético, por lo que se le ha atribuido a esta afección la categoría de enfermedad genética.
| Síndrome de Joubert | ||
|---|---|---|
 | ||
| Especialidad | genética médica | |
Síntomas y anomalías
Principalmente se caracteriza por presentar hipotonía o pérdida del tono muscular, hiperapnea (a veces acompañada de apnea del sueño), ataxia troncal, displasia y el movimiento anormal de los ojos.[1][2]
Todo esto, unido a la falta de vermis en el cerebelo provoca un retraso general en el desarrollo, fatiga y la aparición de retrasos mentales de distinto grado. Pueden darse casos de movimientos imitativos (especulares).
A veces aparecen otro tipo de anomalías menos comunes, como la hipersensibilidad al ruido, polidactilia, meningoencefaloceles, quistes renales o microcefalia.
Este síndrome se ha asociado muy a menudo con el autismo, ya que muchos pacientes diagnosticados muestran criterios para designarlos como pacientes de autismo.
Lo más importante es que los síntomas sólo se muestran en los primeros años del paciente. La persona puede nacer con el trastorno genético, pero si no muestra los síntomas a los pocos meses o años, la enfermedad remitirá y será un individuo sano. Si por el contrario los síntomas permanecen, podrá tratarse, pero la afección será definitiva.
Factores genéticos
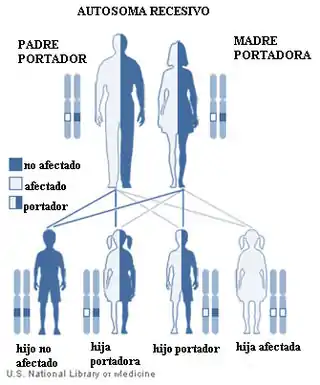
Hasta la fecha, se conocen variantes patogénicas causantes del síndrome de Joubert en 34 genes. En 33 de estos genes se da una herencia autosómica recesiva, mientras que en casos más raros se da una herencia ligada al cromosoma X (en variantes patogénicas que afectan al gen OFD1). Variantes patogénicas en los genes relacionados con este síndrome incluyen pequeñas inserciones o deleciones, mutaciones de cambio de sentido, mutaciones sin sentido y variantes alternativas de corte y empalme.[3]
Por el momento, la anomalía se ha ligado a una deleción del cromosoma 9q 34 el cromosoma se coloca más pequeño o es más algunas veces desaparece (encontrada en una familia consanguínea árabe) y del cromosoma 17p 12 (encontrada en un paciente que también asociaba en síndrome de Smith-Magenis al de Joubert). Aun así, los investigadores no se han puesto de acuerdo en la consecución de genes y loci asociados al síndrome de Joubert.[4][5]
Últimos estudios asocian el gen Arl13b, que sintetiza una proteína implicada en la formación (y probablemente función) de los cilios primarios de las células del cerebelo. La proteína es necesaria para el correcto funcionamiento de un sistema de transporte de proteínas en los cilios primarios.
Otros genes causantes de esta enfermedad son el NPHP1, AHI1, CEP290[6] y INPP5E.[7]
Diagnóstico
Principalmente se basa en datos radiológicos y clínicos. Mediante una resonancia magnética se puede observar la ausencia o falta de la vermis cerebelosa, ausencia del paso de los cilindroejes de un hemisferio cerebral a otro (decusación piramidal) y presentar la fosa posterior normal o disminuida.
De forma histológica es frecuente encontrar agenesia de la vermis, displasia en la unión pontomesencefálica y del bulbo raquídeo o fragmentación de varios núcleos del mesencéfalo.
A la hora de realizar un diagnóstico diferencial, se utilizan como base otras afecciones que presenten ausencia de vermis, como el malformación de Dandy Walker, síndrome orofaciodigital tipo 11, síndrome orofaciodigital tipo IV, síndrome de Senior-Løken.
La detección de mutaciones (como deleciones o duplicaciones) en genes relacionados con el síndrome de Joubert se basa en el uso de técnicas moleculares. Debido a la gran heterogeneidad de las mutaciones que causan este síndrome, estas pruebas de diagnóstico molecular pueden incluir pruebas enfocadas en los genes afectados, en las que se analizan las secuencias de estos para encontrar posibles mutaciones, y también pueden incluir pruebas genómicas como la secuenciación de exomas o del genoma.[3]
El síndrome en la actualidad
El síndrome de Joubert es una de esas enfermedades considerada rara, es decir, con pocos casos diagnosticados. El estudio referente a ella es muy escaso y no existe tratamiento curativo.[8]
Referencias
- Joubert M, Eisenring JJ, Robb JP, Andermann F (septiembre de 1969). «Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation». Neurology 19 (9): 813-25. PMID 5816874.
- Saraiva, JM, Baraitser M (1992) Joubert syndrome: a review. Am. J. Med. Genet. 43: 726-731
- Parisi, Melissa; Glass, Ian (1993). Adam, Margaret P., ed. Joubert Syndrome. University of Washington, Seattle. Consultado el 4 de febrero de 2022.
- Ferland R. J. et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nature Genetics, septiembre de 2004, 36:1008-1013.
- Dixon-Salazar T. et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. "American Journal of Human Genetics", diciembre de 2004, 75(6):979-87.
- Traboulsi EI, Koenekoop R, Stone EM (2006). «Lumpers or splitters? The role of molecular diagnosis in Leber congenital amaurosis». Ophthalmic Genet. 27 (4): 113-5. PMID 17148037. doi:10.1080/13816810601013146.
- Parisi M. A. et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. American Journal of Human Genetics, julio de 2004, 75:82-91.
- Badano, Jose L.; Norimasa Mitsuma, Phil L. Beales, Nicholas Katsanis (septiembre de 2006). «The Ciliopathies : An Emerging Class of Human Genetic Disorders». Annual Review of Genomics and Human Genetics 7: 125-148. doi:10.1146/annurev.genom.7.080505.115610. Consultado el 15 de junio de 2008.
Enlaces externos
- https://web.archive.org/web/20060904204425/http://www.enfermedades-raras.org/es/default.htm
- http://www.eurordis.org/
- http://www.joubertfoundation.com/
- NINDS sitio del síndrome de Joubert Archivado el 6 de septiembre de 2008 en Wayback Machine.
- Investigadores Identifican los Genes del Síndrome de Joubert Archivado el 4 de julio de 2016 en Wayback Machine.
- GeneReviews: síndrome de Joubert