Épilepsie rolandique
L'épilepsie rolandique (bénigne) ou épilepsie (familiale) bénigne (de l'enfant) à pointes centrotemporales (EBPCT) est un syndrome d'épilepsie partielle d'origine génétique à transmission autosomique dominante.
| Spécialité | Neurologie |
|---|
| CIM-10 | G40.0 |
|---|---|
| OMIM | 117100 |
| DiseasesDB | 33998 |
| eMedicine | 1181649 |
| MeSH | D019305 |
![]() Mise en garde médicale
Mise en garde médicale
C'est le plus courant des syndromes d'épilepsie de l'enfant[1]. Les manifestations commencent généralement à l'âge de 3 à 13 ans (principalement entre 5 ou 8 ans)[2] et cessent entre 14 et 18 ans, d'où son caractère « bénin »[3],[4].
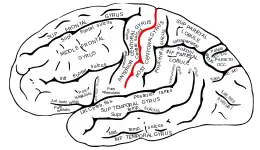
Les crises, parfois appelées crises sylviennes, commencent dans la zone centrotemporale située autour du sillon central du cerveau (également appelé scissure de Rolando, d'après l'anatomiste Luigi Rolando)[5]. Elles se caractérisent par une paresthésie et une activité tonique ou clonique de la partie inférieure du visage, associées à des écoulements de salive et à de la dysarthrie.
Les épisodes surviennent principalement pendant la nuit ; parfois, une généralisation secondaire peut suivre. Dans la plupart des cas, les enfants ont un développement normal.
Signes et symptômes
L'épilepsie rolandique bénigne se caractérise par des attaques focales associées à des signes moteurs et des symptômes somatosensoriels croissants pouvant parfois se généraliser. Entre les crises, l’électroencéphalogramme montre une activité de fond normale, avec des pointes centrotemporales plus marquées au cours du sommeil[6].
Les crises focales, peu fréquentes, souvent uniques, consistent en[7],[8],[9],[10],[11],[12] :
- des symptômes sensorimoteurs faciaux unilatéraux (30 % des patients) ;
- des manifestations oropharyngolaryngées (53 % des patients) ;
- l'impossibilité de parler (40 % des patients) ;
- une hypersalivation (30 % des patients).
Les crises hémifaciales sensorimotrices sont souvent entièrement localisées au niveau de la lèvre inférieure ou se propagent à la main homolatérale. Les manifestations motrices sont des contractions cloniques soudaines, continues ou par à-coups, qui durent généralement de quelques secondes à une minute. Une déviation tonique homolatérale de la bouche est également courante. Les symptômes sensoriels hémifaciaux consistent en un engourdissement unilatéral principalement dans le coin de la bouche. Les crises hémifaciales sont souvent associées à une incapacité à parler et à une hypersalivation : « le côté gauche de ma bouche était engourdi et a commencé à trembler et à tirer vers la gauche, et je ne pouvais pas parler pour dire ce qui m'arrivait ». Une myoclonie négative peut être observée dans certains cas, comme une interruption de l'activité musculaire tonique.
Les manifestations ictales oropharyngolaryngées sont des symptômes sensorimoteurs unilatéraux à l'intérieur de la bouche. L'engourdissement, et plus fréquemment les paresthésies (picotements, picotements, freezing), sont généralement diffus d'un côté ou, exceptionnellement, peuvent être très localisés même sur une dent. Les symptômes moteurs oropharyngolaryngés produisent des sons étranges, évoquant le râle d'agonie, un gargarisme, un grognement ou des sons gutturaux, ainsi que leurs combinaisons : « dans son sommeil, il émettait des bruits gutturaux, la bouche tirée vers la droite, comme s'il mâchait sa langue. Nous l'avons entendue faire des bruits étranges comme un rugissement et l'avons trouvée inconsciente, la tête levée de l'oreiller, les yeux grands ouverts, des torrents de salive sortant de sa bouche, raide. »
L'impossibilité de parler est une forme d'anarthrie. L'enfant est incapable de prononcer un seul mot intelligible et tente de communiquer par des gestes : « Ma bouche s'est ouverte et je ne pouvais pas parler. Je voulais dire que je ne peux pas parler. En même temps, c'était comme si quelqu'un m'étranglait. »
L'hypersalivation, une manifestation autonome importante, est souvent associée à des crises hémifaciales, des symptômes oropharyngolaryngés et l'impossibilité de parler. L'hypersalivation, ce n'est pas seulement de la mousse : « tout à coup ma bouche est pleine de salive, elle coule comme une rivière et je ne peux plus parler. »
Des crises d'épilepsie de type syncope peuvent survenir, probablement un symptôme concomitant du syndrome de Panayiotopoulos : « elle est allongée là, inconsciente, sans mouvements, sans convulsions, comme une poupée de cire, sans vie. »
Conscience et mémoire sont pleinement conservées dans plus de la moitié des cas (58 %) : « J'ai senti que l'air était forcé dans ma bouche, je ne pouvais pas parler et je ne pouvais pas fermer la bouche. Je pouvais bien comprendre tout ce qu'on me disait. D'autres fois, j'ai l'impression qu'il y a de la nourriture dans ma bouche et qu'il y a aussi beaucoup de salive. Je ne peux pas parler. » Dans les autres cas (42 %), la conscience s'altère au cours de la progression ictale et dans un tiers des cas le patient n'a aucun souvenir de la crise.
Une évolution vers des hémiconvulsions ou des crises tonicocloniques généralisées survient chez environ la moitié des enfants et les hémiconvulsions peuvent être suivies d’une hémiparésie post-critique de Todd.
Durée et distribution circadienne : les crises rolandiques sont généralement brèves, d'une durée de 1 à 3 minutes. Les trois quarts des crises surviennent pendant le sommeil à mouvements oculaires non rapides, principalement à l'endormissement ou juste avant le réveil.
État de mal épileptique : bien que rare, un état moteur focal ou un état de mal épileptique hémiconvulsif est plus fréquent que l'état de mal épileptique convulsif secondairement généralisé, qui reste exceptionnel[13],[14]. L'état de mal épileptique operculaire survient généralement chez les enfants ayant une évolution atypique ou peut être induit par la carbamazépine ou la lamotrigine. Cet état dure de plusieurs heures à plusieurs mois et consiste en des contractions unilatérales ou bilatérales continues de la bouche, de la langue ou des paupières, une subtile myoclonie, périorale ou non, positive ou négative, une dysarthrie, un empêchement de la parole, des difficultés à avaler, une apraxie buccofaciale et une hypersalivation. Ceux-ci sont souvent associés à des pics et des vagues incessants sur un EEG pendant le sommeil à mouvements des yeux non rapides.
Autres types de crises : malgré une hypersalivation importante, les crises focales avec des manifestations principalement autonomes (crises autonomes) ne sont pas considérées comme faisant partie du syndrome clinique de base de l'épilepsie rolandique. Cependant, certains enfants peuvent présenter des crises autonomes indépendantes ou des crises mixtes rolandico-autonomes, y compris des vomissements comme dans le cas du syndrome de Panayiotopoulos[15],[16],[17].
Formes atypiques : des manifestations atypiques sont documentées, comme un âge d'apparition de la maladie précoce, un retard de développement ou des difficultés d'apprentissage à l'inclusion, d'autres types de crises ou des EEG atypiques[14],[18],[19],[20].
Ces enfants ont généralement une intelligence et un développement normaux[3]. L'apprentissage n'est pas altéré par l'épilepsie rolandique.
Épidémiologie
L'épilepsie rolandique est le plus courant des syndromes d'épilepsie de l'enfant. Elle en représente 8 à 25 % des cas.
L'âge d'apparition des symptômes varie de 1 à 14 ans, 75 % commençant entre 7 et 10 ans avec une prédominance masculine de 1,5[réf. souhaitée]. Son incidence est estimée à un cas pour 5 000 enfants de la classe d'age concernée (jusqu'à 15 ans)[2].
Causes
L'épilepsie rolandique est considérée être une maladie génétique à transmission autosomique dominante avec dépendance à l'âge et pénétrance variable, bien que toutes les études ne soutiennent pas cette thèse[21],[4],[22],[23]. Les causes seraient multifactorielles[24]. Des études de liaison génétique ont mis en évidence une région de susceptibilité possible sur le chromosome 15q14, à proximité de la sous-unité alpha-7 du récepteur de l'acétylcholine[25]. La plupart des études montrent une légère prédominance masculine[4]. En raison de l'évolution bénigne et de l'occurrence spécifique à l'âge, on pense qu'elle représente une altération héréditaire de la maturation cérébrale[4].
Une association avec le gène ELP4 a été identifiée[26],[27].
Diagnostic
Le diagnostic peut être confirmé lorsque des pics centrotemporaux caractéristiques sont observés par électroencéphalographie (EEG)[28]. En règle générale, on observe des pics intenses suivis d'ondes lentes[29], activés par le sommeil[2]. Compte tenu de l'activité nocturne, un EEG de sommeil est souvent utile. L'imagerie par résonance magnétique cérébrale (IRM) reste normale[2]. Techniquement, la bénignité ne peut être confirmée que si le développement de l'enfant continue d'être normal au cours du suivi[4].
Le diagnostic différentiel inclut d'autres épilepsies idiopathiques focales de l'enfance : épilepsie occipitale bénigne de l'enfance, de type Panayiotopoulos ou Gastaut[2].
Les autres étiologies provoquant des symptômes similaires sont exclues par neuroimagerie — généralement une IRM cérébrale — qui n'est conseillée que pour les cas présentant des manifestations ou des résultats atypiques à l'examen clinique ou à l'EEG. Le trouble doit être distingué de plusieurs autres affections, en particulier les pointes centrotemporales sans crises, les pointes centrotemporales avec pathologie cérébrale locale, les pointes centrales dans le syndrome de Rett ou le syndrome de l'X fragile, l'épilepsie rolandico-sylvienne maligne, l'épilepsie temporale ou le syndrome de Landau-Kleffner.
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Rolandic epilepsy » (voir la liste des auteurs).
- Kramer U, « Atypical presentations of benign childhood epilepsy with centrotemporal spikes: a review », J. Child Neurol., vol. 23, no 7, , p. 785–90 (PMID 18658078, DOI 10.1177/0883073808316363)
- Orphanet — Rolandic epilepsy.
- Wirrell EC, « Benign epilepsy of childhood with centrotemporal spikes », Epilepsia, vol. 39 Suppl 4, , S32–41 (PMID 9637591, DOI 10.1111/j.1528-1157.1998.tb05123.x)
- Chahine LM, Mikati MA, « Benign pediatric localization-related epilepsies », Epileptic Disord, vol. 8, no 4, , p. 243–58 (PMID 17150437, lire en ligne)
- Benign Rolandic epilepsy. Retrieved August 8, 2008.
- Épilepsie rolandique — Descripteur MeSH
- Marc Beaussart, « Benign epilepsy of children with Rolandic (centro-temporal) paroxysmal foci. A clinical entity. Study of 221 cases. », Epilepsia, vol. 13, no 6, , p. 795–811 (PMID 4509173, DOI 10.1111/j.1528-1157.1972.tb05164.x)
- P Loiseau et Beaussart, M, « The seizures of benign childhood epilepsy with Rolandic paroxysmal discharges. », Epilepsia, vol. 14, no 4, , p. 381–389 (PMID 4521094, DOI 10.1111/j.1528-1157.1973.tb03977.x)
- P Lerman et Kivity, S, « Benign focal epilepsy of childhood. A follow-up study of 100 recovered patients. », Archives of Neurology, vol. 32, no 4, , p. 261–264 (PMID 804895, DOI 10.1001/archneur.1975.00490460077010)
- Chrysostomos P. Panayiotopoulos, Benign Childhood Partial Seizures and Related Epileptic Syndromes, Londres, John Libbey Eurotext, , 33–100 p. (ISBN 978-0-86196-577-9, lire en ligne), « Benign childhood epilepsy with centrotemporal spikes or Rolandic seizures »
- Bernardina Dalla, Vincenzo Sgro et Natalio Fejerman, Epileptic Syndromes in Infancy, Childhood, and Adolescence, France, John Libbey Eurotext, , 203–225 p. (ISBN 978-2-7420-0569-7), « Epilepsy with centro-temporal spikes and related syndromes »
- C. P. Panayiotopoulos, Michael, M., Sanders, S., Valeta, T. et Koutroumanidis, M., « Benign childhood focal epilepsies: assessment of established and newly recognized syndromes », Brain, vol. 131, no 9, , p. 2264–2286 (PMID 18718967, DOI 10.1093/brain/awn162
 )
) - Deonna T, Ziegler AL, Despland PA, « Combined myoclonic-astatic and "benign" focal epilepsy of childhood ("atypical benign partial epilepsy of childhood"). A separate syndrome? », Neuropediatrics, vol. 17, no 3, , p. 144–51 (PMID 3762871, DOI 10.1055/s-2008-1052516)
- Natalio Fejerman, Caraballo, Roberto et Tenembaum, Silvia N., « Atypical Evolutions of Benign Localization-Related Epilepsies in Children: Are They Predictable? », Epilepsia, vol. 41, no 4, , p. 380–390 (PMID 10756401, DOI 10.1111/j.1528-1157.2000.tb00177.x)
- Michalis Koutroumanidis et Chrysostomos P. Panayiotopoulos, « Chapter 9: Benign childhood seizure susceptibility syndromes », ILAE, (lire en ligne) in ILAE - Epilepsy 2017
- Caraballo R, Cersosimo R, Fejerman N, « Panayiotopoulos syndrome: a prospective study of 192 patients », Epilepsia, vol. 48, no 6, , p. 1054–61 (PMID 17442007, DOI 10.1111/j.1528-1167.2007.01085.x)
- Specchio N, Trivisano M, Di C, Cappelletti S, Masciarelli G, Volkov J, etal, « Panayiotopoulos syndrome: A clinical, EEG, and neuropsychological study of 93 consecutive patients », Epilepsia, vol. 51, no 10, , p. 2098–107 (PMID 20528983, DOI 10.1111/j.1528-1167.2010.02639.x)
- Datta A, Sinclair DB, « Benign epilepsy of childhood with rolandic spikes: typical and atypical variants », Pediatr Neurol, vol. 36, no 3, , p. 141–5 (PMID 17352945, DOI 10.1016/j.pediatrneurol.2006.12.003)
- Kramer U, « Atypical presentations of benign childhood epilepsy with centrotemporal spikes: a review », J Child Neurol, vol. 23, no 7, , p. 785–90 (PMID 18658078, DOI 10.1177/0883073808316363)
- Wirrell EC, Camfield PR, Gordon KE, Dooley JM, Camfield CS, « Benign rolandic epilepsy: atypical features are very common », Journal of Child Neurology, vol. 10, no 6, , p. 455–8 (PMID 8576555, DOI 10.1177/088307389501000606)
- http://association.gens.free.fr/NEUROLOGIA/EMC%20neurologie/1%20Genetique/$Aspects%20genetiques%20des%20epilepsies%20EMC.pdf EMC — Aspects génétiques des épilepsies
- Neubauer BA, « The genetics of rolandic epilepsy », Epileptic Disord, vol. 2 Suppl 1, , S67–8 (PMID 11231229, lire en ligne)
- Bali B, Kull LL, Strug LJ, etal, « Autosomal dominant inheritance of centrotemporal sharp waves in rolandic epilepsy families », Epilepsia, vol. 48, no 12, , p. 2266–72 (PMID 17662063, PMCID 2150739, DOI 10.1111/j.1528-1167.2007.01221.x)
- Caractéristiques génétiques de l’épilepsie.
- Neubauer BA, Fiedler B, Himmelein B, etal, « Centrotemporal spikes in families with rolandic epilepsy: linkage to chromosome 15q14 », Neurology, vol. 51, no 6, , p. 1608–12 (PMID 9855510, DOI 10.1212/WNL.51.6.1608)
- Une équipe internationale de chercheurs, menée par le Dr Pal de l’université de Columbia, vient de mettre au jour le premier gène lié à la forme d’épilepsie la plus commune, l’épilepsie rolandique familiale.
- Strug LJ, Clarke T, Chiang T, Modèle:Etal., « Centrotemporal sharp wave EEG trait in rolandic epilepsy maps to Elongator Protein Complex 4 (ELP4) », Eur. J. Hum. Genet., vol. 17, no 9, , p. 1171–1181 (PMID 19172991, PMCID 2729813, DOI 10.1038/ejhg.2008.267)
- Blueprints Neurology, 2nd ed.
- Stephani U, « Typical semiology of benign childhood epilepsy with centrotemporal spikes (BCECTS) », Epileptic Disord, vol. 2 Suppl 1, , S3–4 (PMID 11231216, lire en ligne)
Voir aussi
Personnalités affectées
Liens externes
 Portail de la médecine
Portail de la médecine  Portail de la psychologie
Portail de la psychologie  Portail des neurosciences
Portail des neurosciences