Étude d'association pangénomique
Une étude d'association pangénomique (en anglais genome-wide association study, GWAS) est une analyse de nombreuses variations génétiques chez de nombreux individus, afin d'étudier leurs corrélations avec des traits phénotypiques[1].
Ces études se concentrent généralement sur les associations entre les polymorphismes nucléotidiques (SNP) et des phénotypes tels que les maladies humaines majeures.
En effet, quand elle est appliquée sur des données humaines, une comparaison de séquences d’ADN se fait entre individus ayant plusieurs phénotypes différents pour un même caractère, la taille par exemple. Cette comparaison a pour but d’identifier les variants génétiques les plus fréquemment associés avec le phénotype observé, on parle alors d’une approche « phénotype en premier ». Cette approche consiste à catégoriser les patients selon leur signes cliniques, et de les corréler avec des SNP à l’inverse de l’approche « génotype en premier ».
Par exemple, l'étude d'association pangénomique « CoLaus » (cohorte lausannoise) cherche des associations entre les données médicales et génétiques d'une cohorte de plus de 6 000 personnes[2],[3],[4]. Les échantillons d’ADN de chaque personne sont alors collectés et déposés sur des puces à ADN contenant des millions de sondes auxquelles les variants génétiques vont s’hybrider et être détectés par fluorescence.
Une GWAS est appliquée sur l’entièreté du génome, ce qui permet d’obtenir un pourcentage plus élevé de variance expliquée pour des traits quantitatifs contrôlés par plusieurs gènes. Néanmoins, les GWAS ne donnent pas une relation de causalité du SNP à son caractère mais renseignent plutôt sur les variants corrélés avec ce trait[5],[6],[7].
Méthodologie
Pour faire une étude d’association pangénomique il faut disposer d'une cohorte, c’est-à-dire des milliers d'individus à partir desquels de l’ADN pourra être collecté. L'approche la plus commune est l’Étude cas-témoins, qui compare deux grands groupes d'individus, un groupe témoin en bonne santé et un groupe de cas affecté par une maladie. Il faut également disposer de marqueurs génétiques, ici des SNPs connus. La première étape est le génotypage des échantillons d'ADN de la cohorte afin de déterminer des variants génétiques. Cela peut être fait par une technologie appelée SNP array. Ensuite, les sujets de la cohorte sont suivis au fil du temps et subissent régulièrement des analyses au niveau des phénotypes important pour l’analyse. Le style de vie des sujets de la cohorte peut aussi être pris en compte pour étudier l’influence des facteurs environnementaux.
Pour chacun de ces SNP, il est étudié si la fréquence de l'allèle est significativement modifiée entre le groupe cas et le groupe témoin. Ceci est calculé grâce à un Odds ratio qui est le rapport de deux cotes, qui sont les chances de maladie pour les individus ayant un allèle spécifique et les chances de maladie pour les individus qui n'ont pas ce même allèle. Lorsque la fréquence de l'allèle dans le groupe cas est beaucoup plus élevée que dans le groupe témoin, le rapport de cotes est supérieur à 1, et vice versa. De plus, une valeur de P pour la significativité du odds-ratio est typiquement calculée en utilisant un Test du χ². Trouver des odds ratios significativement différents de 1 est l'objectif de l'étude d’association pangénomique parce que cela montre qu'un SNP est associé à une maladie.
Résultats
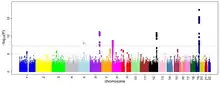
On peut représenter les résultats d’une GWAS sur un graphique appelé diagramme de Manhattan. Il s’agit d’un nuage de points (statistique), avec les coordonnées génomiques affichées le long de l'axe X et le logarithme négatif de la valeur P de l'association pour chaque polymorphisme nucléotidique (SNP) affiché sur l'axe Y. Ainsi chaque point du diagramme de Manhattan représente un SNP. Du fait que les associations les plus fortes ont les plus petites valeurs de P leurs logarithmes négatifs seront les plus grands. Donc lorsque la valeur p est très petite (significative), le point sera très élevé. Étant donné que plusieurs tests sont effectués, il faut ajuster le seuil de significativité avec une correction de Bonferroni par exemple.
Limitations
L’étude d’association pangénomique comprend plusieurs limitations principalement au niveau statistique : taille de l’échantillon, nombre de tests utilisés, stratification de la population (fréquences alléliques différentes au sein d’une même population), tous constituent un facteur pour lequel il faut contrôler afin d’avoir des résultats interprétables[8]. Aussi, les traits analysés sont souvent polygéniques avec une petite taille d'effet (Effect size en anglais), donc l’augmentation de la taille de l’échantillon permettrait une augmentation de la puissance statistique[8].
Les critiques principales des études d’association pangénomique, ciblent surtout la supposition de ces dernières que des allèles communs expliquent une bonne partie de l’héritabilité génétique des maladies humaines[9]. Or les résultats empiriques réfutent cette hypothèse et montrent que pour chaque variant associé il y a une petite augmentation du risque de la maladie[10]. Néanmoins, certaines méta-analyses ont proposé des solutions combinant GWAS et déséquilibre de liaison pour augmenter le pourcentage de variance expliquée[11].
Notes et références
- (en) Laura Sanders, « 40 more ‘intelligence’ genes found; Evidence grows for the idea that some of your smarts are in your DNA », sur Science News, (consulté le )
- Site de l'étude CoLaus (page consultée le 29 décembre 2011).
- Philippe Barraud, « Maladies cardio-vasculaires : 6000 Lausannois sous la loupe d'une vaste étude au CHUV », Le Temps, mercredi 10 septembre 2003 (article consulté en ligne le 29 décembre 2011).
- Pierre-Yves Frei, « 7000 Romands sous la loupe médicale d’une vaste étude génétique », Le Temps, mardi novembre 2011 (article consulté en ligne le 29 décembre 2011).
- (en) Teri A. Manolio, « Genomewide Association Studies and Assessment of the Risk of Disease », New England Journal of Medicine, vol. 363, no 2, , p. 166–176 (ISSN 0028-4793 et 1533-4406, DOI 10.1056/nejmra0905980, lire en ligne, consulté le )
- (en) Thomas A. Pearson, « How to Interpret a Genome-wide Association Study », JAMA, vol. 299, no 11, , p. 1335 (ISSN 0098-7484, DOI 10.1001/jama.299.11.1335, lire en ligne, consulté le )
- (en-US) « Genome-Wide Association Studies Fact Sheet », sur National Human Genome Research Institute (NHGRI) (consulté le )
- Arthur Korte et Ashley Farlow, « The advantages and limitations of trait analysis with GWAS: a review », Plant Methods, vol. 9, , p. 29 (ISSN 1746-4811, PMID 23876160, PMCID PMC3750305, DOI 10.1186/1746-4811-9-29, lire en ligne, consulté le )
- Peter M. Visscher, Matthew A. Brown, Mark I. McCarthy et Jian Yang, « Five years of GWAS discovery », American Journal of Human Genetics, vol. 90, no 1, , p. 7–24 (ISSN 1537-6605, PMID 22243964, PMCID PMC3257326, DOI 10.1016/j.ajhg.2011.11.029, lire en ligne, consulté le )
- Carl A. Anderson, Nicole Soranzo, Eleftheria Zeggini et Jeffrey C. Barrett, « Synthetic associations are unlikely to account for many common disease genome-wide association signals », PLoS biology, vol. 9, no 1, , e1000580 (ISSN 1545-7885, PMID 21267062, PMCID PMC3022527, DOI 10.1371/journal.pbio.1000580, lire en ligne, consulté le )
- (en) Jian Yang, Teresa Ferreira, Andrew P Morris et Sarah E Medland, « Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits », Nature Genetics, vol. 44, no 4, , p. 369–375 (ISSN 1061-4036 et 1546-1718, PMID 22426310, PMCID PMC3593158, DOI 10.1038/ng.2213, lire en ligne, consulté le )