Spectrométrie d'absorption des rayons X
La spectrométrie d'absorption des rayons X est une spectrométrie d'absorption qui aide à déterminer la structure d'un matériau. Elle a l'avantage d'être sélective quant à l'espèce atomique observée.
Histoire
Au début du 20e siècle, la technique de spectroscopie d’absorption des rayons X (XAS) (SAX) n’était pas assez avancée pour représenter le futur de la science. Son avancement est surtout dû à des chercheurs avec énormément de motivation, mais n’ayant pas les capacités d’expliquer la théorie derrière un phénomène.
Les débuts de la spectroscopie d’absorption des rayons X ont commencé en l’an 1913. Cette découverte est attribuée à Maurice de Broglie grâce à son expérience. Celle-ci se compose d’un cristal sur un cylindre de baromètre enregistreur. Le cristal peut tourner à vitesse angulaire constante autour de l’axe vertical grâce à un mécanisme d’horloge intégré. L’observation de Broglie est que l’énergie du rayon X résultant varie en fonction de l’angle du cristal entre l’angle du rayon incident et l’angle critique. Ces énergies ont été enregistrées sur une plaque photographique qui contenait du bromure d’argent AgBr, ce qui correspondait également avec le spectre d’absorption de l’argent et du brome[1].
En 1916, une grande avancée dans la spectroscopie d’absorption a vu le jour. Manne Siegbahn et Stenström ont développé le premier spectromètre à rayons X sous vide, réduisant au maximum les interférences de l’air. Grâce à cette invention, les premiers spectres d’absorption expérimentaux ont pu être découverts par Fricke en 1920 pour les pics-K des éléments du magnésium au chrome et par Hertz en 1921 pour les pics-L des éléments du césium au néodyme[1].
C’est en 1931 que Hanawalt remarque que les états chimiques et physiques de l’échantillon affectent la structure du spectre puisque les solides se subliment en présence de rayons X.
Au courant des années 1960, l’équipement du spectromètre d’absorption est amélioré pour obtenir de meilleurs spectres. Un cristal en silice agit comme monochromateur et est placé sur un goniomètre pour capter tous les angles de déviation des rayons X[1].
En 1971, ce sont les scientifiques Sayers, Stern et Lytle qui ont fait une percée majeure dans l’interprétation des oscillations post-pics. En faisant une transformée de Fourier sur la section comportant les oscillations, ils ont remarqué une distribution radicalaire de la densité atomique[1].
Au cours de ces années, la spectroscopie d’absorption des rayons X a été améliorée par l’existence d’une solide théorie permettant l’analyse quantitative de spectres XAS, l’accès à des sources de rayonnement plus fiables faisant par la même occasion augmenter la qualité des spectres XAS et le développement d’une banque de codes comportant les données d’analyse dans une voie sûre, reproductible et contrôlée[1].
Matériel, montage et instruments

La plupart des expérimentations en spectrométrie d'absorption des rayons X (XAS) sont faites à partir d’une radiation de rayons x émise par un synchrotron ou par des tubes à rayons X. Le synchrotron fournit une gamme d’énergie de rayons X permettant d’analyser la plupart des éléments du tableau périodique[2].
Depuis plusieurs décennies, les tubes à rayons X sont une bonne source de photons à rayons X. Ils utilisent une haute tension entre la cathode et l'anode, ce qui produit un courant d'électrons qui crée un impact à l'anode (Bremsstrahlung), ce qui produit une large distribution de rayons X. Comme la plupart de l'énergie utilisée pour le courant électronique est transférée en chaleur, seulement 1 % de l'énergie sert à la production des rayons X[3]. La limite d'intensité de rayons X est déterminée par le point de fusion de l'anode, donc les tubes à rayons X à micro-focalisation sont à surveiller, car ils génèrent un chauffage intense. Une technologie de tubes anodiques à liquide métalliques permettent un chauffage plus intense. De plus, l'émission de rayonnement X des tubes est divergente : les rayons s'éloignent de la source, un collimateur est donc nécessaire pour pouvoir optimiser l'utilisation des rayons X, sinon, seulement une petite partie du rayonnement peut être utilisée[3].
L'autre source de rayons X utilisée est le synchrotron. La première partie du synchrotron est la source d’électrons : généralement un synchrotron avec laser à électrons libres (X-FELs)[4]. Les électrons sont accélérés dans un accélérateur linéaire (LINAC) avec un vide poussé avant que leurs énergies soient encore plus augmentées dans un anneau d’accélération par des aimants de courbure puissants. Les électrons sont ensuite transférés dans l’anneau de stockage où ils circulent à une vitesse très élevée. En fait, ils font plus d'un million de tours par seconde, ce qui crée des radiations électromagnétiques intenses. La ligne de faisceaux envoie ensuite la radiation en direction de l’expérience. Des miroirs sont utilisés pour diriger le rayonnement tandis que des fentes réduisent la taille du faisceau. Un monochromateur ou un système de filtres permet de choisir l’énergie des rayons X avec précision[5]. Les deux types de monochromateurs principalement utilisés avec les rayons X sont les monochromateurs à réseau et les monochromateurs cristallins. Les monochromateurs à réseau utilisent soit un réseau plat (avec des miroirs incurvés : le miroir de focalisation et le collimateur, pour faire la mise au point) ou un réseau courbé pour réfléchir le rayon selon les longueurs d’onde de ses composantes. Des ajustements de l’entrée, du réseau et de la sortie permettent alors de sélectionner la longueur d’onde désirée. Une simple rotation du détecteur autour du cercle de Rowland pour les réseaux courbés suffit à sélectionner la longueur d'onde désirée. Les monochromateurs cristallins fonctionnent avec à des longueurs d’onde plus basses selon le principe de diffraction de Bragg avec un ou deux cristaux de haute qualité. Les cristaux s’ajustent en rotation pour permettre le contrôle de l’énergie du photon[6]. Après le passage du faisceau de rayon X à travers l’échantillon, l’intensité du faisceau émergent doit être mesurée avec un détecteur de rayons X. La plupart des détecteurs de rayons X utilisent un mécanisme à deux étapes : la transformation des rayons X en électrons, en photons visibles, en paire de trous d’électrons pour un semiconducteur ou en paire d’ions pour un gaz. C’est cette nouvelle particule qui sera détectée[7].
Principe d'absorption de rayons X et excitation de l'atome
Dans le spectre des rayonnements, on classe les rayons X dans la gamme d’ondes qui ont une longueur d’onde entre 3 nanomètres et 0.03 nanomètres[8]. En référence, le spectre des ondes visibles varie de 400 à 700 nanomètres. On peut mesurer l’énergie d’un rayon X selon l’équation suivante :

Où h = constante de planck, c = vitesse de la lumière et λ = longueur de l’onde
On représente alors cette énergie en kilo électron-volts (keV). Cette même unité d’énergie est utilisée pour définir la force des liaisons entre atomes, on voit aussi que les rayons X se retrouvent dans trois ordres de grandeur, soit de 10−9 à 10−11 mètres, et que la grandeur des rayons atomiques se situe dans les angströms[9] (1 Å = 10−10 mètres) Le rayon atomique ici désigne plutôt la distance entre le centre du noyau et l’extrémité de la plus grande orbitale. Les rayons x étant dans la même grandeur que les rayons atomiques, ils ont plus de facilité à les pénétrer. Le seul type de longueur d’onde plus petit que le rayon x est le rayon gamma. Celui-ci pénètre plutôt dans le noyau. Vu la différence dans les ordres de grandeur des rayons X, l’ordre de grandeur des énergies peut varier de 1 keV à 100 keV[10]. Les rayons à plus grande énergie ont également une longueur d’onde plus petite.

Lorsqu’un rayon X frappe un atome, celui-ci absorbe cette énergie, ce qui lui permet de prendre un électron d’une couche inférieure et l’amener à une couche supérieure[11]. Or le rayon X ne va pas nécessairement toujours exciter un électron d’une même couche basse en énergie pour l’envoyer dans la couche haute en énergie. En tenant compte de la période de l’élément et des configurations électroniques de ceux-ci, ce n’est pas toujours la même couche (ex. n = 1) qui se fait exciter pour passer à la même couche supérieure (ex. n=3, pour les atomes de la troisième période). Après l’excitation d’un électron vers une couche supérieure, il reste un vide d’électron dans une couche interne de l’atome qui se fait remplir par un électron de la prochaine couche. Cela décale les électrons dans l’atome pour s’assurer qu’il n’y a pas de manque d’électron dans aucune des couches internes. Par exemple, si la couche (n =1) se fait exciter et qu’il manque un électron. La couche (n = 2) va la remplir, mais s’il existe une couche (n = 3), celle-ci va remplir la couche (n =2). Il est entendu qu’un électron ne peut pas passer d'une couche inférieure, qui serait plus basse en énergie, vers une couche plus haute en énergie pour combler le vide. Or, un détecteur ne peut pas détecter la quantité de rayons x absorbés (ou le kEV absorbé). Lorsque la couche inférieure, préalablement excitée par le rayon x, retourne à son état stable (et rempli) grâce à la transition d'un électron provenant d'une couche extérieure, de l'énergie est émise à une certaine longueur d'onde sous forme de radiation afin de permettre à l'atome de baisser son niveau d'énergie et de se restabiliser. Il retourne alors à son état initial avant l'excitation[12].
La notation pour ces déplacements d’électrons dépend de quelle couche a été excitée et un électron de quelle couche a été utilisé pour combler le vide dans une couche inférieure. On nomme la première couche (n =1) ‘K’. La deuxième (n=2) ‘L’, la troisième (n=3) ‘M’ et ainsi de suite[13]. Si l’électron d’une couche inférieure a sauté une couche, et donc ne traverse qu’une couche pour revenir, l’énergie émise est notée ‘α’. Si 2 couches sont traversées, ‘β’, etc. Cette notation est basée sur la notation des transitions électroniques (d'absorption et d’émission) de Siegbahn.
Donc, on nomme un déplacement d’un électron qui traverse deux couches pour revenir à sa couche initiale, qui était la couche ‘K’ (n =1), on la désigne par ‘Kβ’. Si l’électron redescend d’une couche pour combler la couche inférieure en manque d’électron ‘L’ (n = 2), on désigne la transition ‘Lα’. Toutes ces transitions électroniques ont des énergies différentes et sont quantifiables.
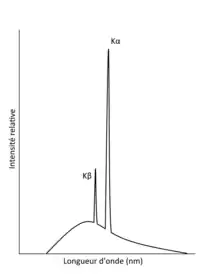
En analyse quantitative, il est possible de mesurer les radiations émises lorsque la couche interne se fait remplir. Des valeurs empiriques pour les ‘Kα’, ‘Kβ’, etc sont connues (en keV)[14]. Ensuite en mesurant les intensités des radiations détectées il est possible d’identifier les atomes en les comparant aux références. Cela permet de distinguer entre deux pics lequel est β et lequel est α, lequel est K, lequel est M, etc[15]. La spectroscopie des rayons x permet depuis ces valeurs de confirmer la structure électronique des atomes et de confirmer leur identité.
Applications analytiques de la spectrométrie de rayons X
Les applications de la spectroscopie d’absorption des rayons X touchent à plusieurs secteurs de la science, que ce soit en laboratoire pour des analyses de routine, en environnement, en géologie ou en industrie.
Catalyse : La XAS permet de suivre la progression d’une réaction catalytique par l’identification des composantes. Les réactions cinétiques sont les premières sources d’utilisation de la XAS lors de l’utilisation d’un catalyseur[16].
Hautes pressions : L’analyse de XAS à pression élevée permet entre autres l’amélioration du signal reçu par le transducteur. Avec des hautes pressions, les éléments agissent comme l’état amorphe et peuvent être comparés[17].
Complexes de coordination : La XAS permet l’observation de la géométrie et de la conformation électronique d’un complexe. Ce processus est possible par le passage d’un électron d’une couche électronique faible à une couche plus élevée. Un pic-K pour un métal est dû à un transfert de la couche 1s à la couche 3d ou 4p alors qu’un pic-L pour un métal est transfert de la couche 2p à la couche 3d[18].
Semiconducteurs : Dans les matériaux semiconducteurs, certaines méthodes chimiques sont utilisées pour améliorer les effets de ces matériaux. Les techniques d’optimisation les plus courantes sont l’utilisation du dopage, l’insertion d’un élément dans la structure d’un matériau, le greffage d’un film sur le matériau et les nanostructures. La XAS est là pour identifier l’action qu’a le composé optimisant pour évaluer les effets de ceux-ci sur un semiconducteur[19].
Oxydes à l’état solide : La XAS a plusieurs utilités pour les composés à l’état solide. Un de ces plus grands secteurs est les oxydes. En effet, les oxydes peuvent avoir plusieurs formes à cause de sa possibilité d’avoir des orbitales vides ou pleines en plus de pouvoir faire des liens covalents. Grâce à la XAS, les angles des oxydes peuvent être observés à cause de la différence de longueur de lien entre les composés. Puisque la longueur des liens peut être observée, il existe aussi certains métaux de transition qui ont des longueurs de liens variant en fonction de leur niveau d’oxydation (ex : Co, Mn)[20].
Nucléaire : L’utilisation principale de la XAS dans l’industrie du nucléaire est la détection de l’uranium dans l’ensemble de sa production. Du côté de la mine, l’uranium peut être détecté directement dans la roche pour cibler les proportions d’uranium brut dans la roche. Un peu plus loin, dans la purification de l’uranium, la XAS sert à déterminer les impuretés pour ensuite les éliminer et ainsi produire du carburant à l’uranium plus pur[21].
Géologie : La XAS dans le domaine de la géologie sert surtout à l’identification des composantes des roches. Cela va de l’étude de la formation des minéraux, la structure cristalline des pierres précieuses jusqu’à l’origine de roches extraterrestres à partir de météorites[22].
Environnement : La XAS a plusieurs utilités dont la remédiation de sites de minage et industriels, le traitement des eaux usées et la phytoremédiation. Tous sont pour l’identification des matières néfastes pour ensuite pouvoir les traiter convenablement[22].
Bien que les applications soient tout de même variées, elles sont tout de même plus limitées lorsqu’elles sont comparées avec des techniques similaires, soit la spectroscopie de fluorescence et d’émission des rayons X. Cette limite est surtout due au fait que les échantillons doivent contenir une matrice ayant le moins d’effet parce que la majorité de l’interférence ce fait par le biais de la matrice[23].
Méthodes complémentaires XANES et EXAFS

Nous savons que la spectroscopie d’absorption de rayons X fait une mesure de la décroissance du faisceau de photons d’une certaine longueur d’onde à travers l’échantillon analysé. Pour avoir un spectre, comme celui présenté ci-contre avec les différentes régions, on fait varier l’énergie des photons pour analyser les éléments choisis[24]. Il est aussi possible d’utiliser les techniques complémentaires EXAFS (Extended X-Ray-Absorption Fine-Structure) pouvant être traduit par « structure fine étendue de l'absorption de rayons X » et XANES (X-Ray Absorption Near Edge Structure), traduit par « structure proche du seuil d'absorption de rayons X ». Les deux méthodes peuvent être utiles pour déterminer l’information structurelle. En effet, les spectres XANES rapportent la structure électronique et la symétrie du site métallique. Les spectres EXAFS rapportent le nombre, les types et les distances entre l’élément absorbant et les ligands ou les atomes voisins[25]. La spectrométrie XANES apporte de l’information sur l’état d’oxydation et sur l’environnement de coordination d’atomes de métaux. À cause de l’effet d’écran, le seuil K d’énergie augmente avec l’augmentation de l’état d’oxydation. En effet, dans un atome avec plusieurs électrons, les électrons externes sont, à la fois, attirés par le noyau positif et repoussés par les autres électrons de charge négative. Plus l’état d’oxydation est élevé, plus l’atome est chargé positivement, il nécessite donc plus d’énergie pour exciter un électron, ce qui augmente le niveau d’énergie du seuil K[26]. La spectrométrie EXAFS permet d’analyser l’absorbance des niveaux d'énergies un peu plus élevés que la LUMO (Lowest Unoccupied Molecular Orbital). Les rayons X permettent d’ioniser l’atome en relâchant un photoélectron. L’excès d’énergie envoyé est transféré en énergie cinétique translationnelle. Les atomes proches ont ainsi une influence sur l’énergie du photoélectron relâché. Il y a des interférences entre l’onde du photoélectron sortant et l’onde diffusée par les atomes voisins. À une certaine énergie, ces ondes sont en phase, ce qui crée une interférence constructive et qui augmente le coefficient d’absorption. À une certaine autre énergie, ces ondes sont hors-phase, ce qui crée une interférence destructive et qui réduit le coefficient d’absorption. Si on fait une transformée de Fourier de la modulation, on peut avoir des informations sur les distances entre l’atome d’absorption et les atomes liés. Par contre, la limite de gamme est de 4 à 5 Å de longueur[27].
Avantages et inconvénients de la méthode
Avantages
Il est possible de sonder des échantillons sans l’ordre à grande distance des cristaux avec la technique XAS. En fait, XAS peut détecter les variations dans l’échantillon total, pas seulement de la partie cristalline, ce qui la distingue la méthode de diffraction de rayons X qui ne peut pas identifier les particules cristallines plus petites que 3nm ou les agglomérations amorphes[28].
Les analyses XAS sur les minéraux ont comme avantage qu’on peut utiliser des échantillons très dilués. Cette caractéristique permet d’analyser le mécanisme de substitution de certains éléments dans les minéraux, même ceux en trace[29].
Comme la technique XAS est spécifique, on peut faire l’analyse d’un seul élément sans l’interférence des autres éléments. Par exemple, dans une protéine, on peut étudier les environnements chimiques des différents métaux sélectivement. De plus, l’étude des molécules biologiques par rayonnement X peut être dommageable pour la molécule à cause des radicaux libres et des électrons hydratés. Comme l’analyse XAS nécessite peu de rayons X par rapport à la cristallographie, on peut mieux contrôler les dommages, en plus d’utiliser un système cryogénique pour réduire la destruction[30].
Inconvénients
La spectrométrie d’absorption des rayons X est assez limitée en comparaison avec la technique d’émission de rayons X et la spectrométrie de fluorescence. Pour pouvoir avoir peu d’effet de matrice lors des expériences d’absorption, on doit recourir à des techniques encombrantes prenant beaucoup plus de temps qu’en fluorescence. C’est pour ces raisons que les échantillons doivent à la base avoir un effet de matrice réduit, ce qui réduit le champ d’application de la méthode[23].
Bibliographie
- (en) Jeroen A. Van Bokhoven et Carlo Lamberti, X-Ray Absorption and X-Ray Emission Spectroscopy : Theory and Applications, Chichester, Wiley, , 890 p. (ISBN 978-1-118-84423-6, OCLC 908287071, lire en ligne)
- (en) Junko Yano et Vittal K. Yachandra, « X-ray absorption spectroscopy », Photosynthesis Research, vol. 102, nos 2-3, (ISSN 0166-8595 et 1573-5079, DOI 10.1007/s11120-009-9473-8, lire en ligne, consulté le )
- (en) Douglas A. Skoog, F. James Holler et Stanley R. Crouch, Principles of Instrumental Analysis, Belmont, Thomson Brooks/cole, , 1039 p. (ISBN 978-0-495-01201-6, OCLC 77224390)
Notes et références
- Van Bokhoven et Lamberti 2016, p. 3-8
- Yano et Yachandra 2009, p. 241
- Van Bokhoven et Lamberti 2016, p. 157
- Van Bokhoven et Lamberti 2016, p. 25
- (en) Claudia S. Schnohr et Mark C. Ridgway, X-ray absorption spectroscopy of semiconductors, Berlin, Heidelberg, Springer, , 361 p. (ISBN 978-3-662-44362-0, OCLC 898138420, lire en ligne), p. 12-14
- Van Bokhoven et Lamberti 2016, p. 43
- Van Bokhoven et Lamberti 2016, p. 47
- (en) Science Mission Directorate, « X-Rays », NASA Science, (consulté le )
- (en) J. C. Slater, « Atomic Radii in Crystals », The Journal of Chemical Physics, vol. 41, no 10, , p. 3199–3204 (DOI 10.1063/1.1725697)
- (en) G. D. Holman, « What are Hard X-Rays?, Goddard Space and Flight Center, NASA. » (consulté le )
- (en) Manne Siegbahn, « X-ray spectroscopy », dans P. P. Ewalds, Fifty Years of X-Ray Diffraction, Springer US, , 733 p. (ISBN 978-1-4615-9963-0, DOI 10.1007/978-1-4615-9961-6)
- (en) R. Nave, « X-Ray Transitions. Quantum Physics » (consulté le )
- (en) A. Thompson, D. Attwood, E. Gullikson, M. Howells, K-J. Kim et al., X-ray data booklet (rev. 3), Lawrence Berkeley National Laboratory, University of California, Berkeley, CA 94720, (lire en ligne)
- (en) R. Nave, « Moseley Plot of Characteristic X-Rays, Quantum Physics. » (consulté le )
- Van Bokhoven et Lamberti 2016, p. 353-374
- Van Bokhoven et Lamberti 2016, p. 385-401
- Van Bokhoven et Lamberti 2016, p. 407-432
- Van Bokhoven et Lamberti 2016, p. 437-455
- Van Bokhoven et Lamberti 2016, p. 459-480
- Van Bokhoven et Lamberti 2016, p. 523-551
- Van Bokhoven et Lamberti 2016, p. 561-599
- Skoog, Holler et Crouch 2007, p. 325
- Van Bokhoven et Lamberti 2016, p. 61
- Yano et Yachandra 2009, p. 242
- Yano et Yachandra 2009, p. 243
- Yano et Yachandra 2009, p. 244
- Van Bokhoven et Lamberti 2016, p. 816
- Van Bokhoven et Lamberti 2016, p. 566
- Yano et Yachandra 2009, p. 246
Voir aussi
Articles connexes
- European Synchrotron Radiation Facility
- Loi de Beer-Lambert
- Théorie de la diffraction sur un cristal
 Portail de la physique
Portail de la physique  Portail de la chimie
Portail de la chimie