Dystrophie myotonique de Steinert
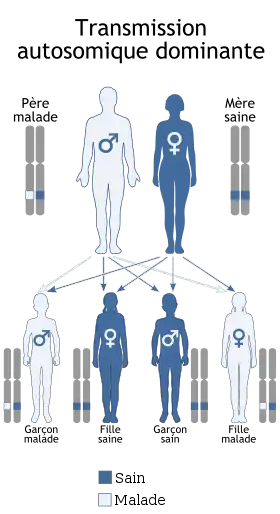
La dystrophie myotonique de Steinert ou maladie de Curschmann-Steinert ou encore dystrophie myotonique de type I (DM1), est une maladie génétique autosomique dominante, à pénétrance incomplète et marquée par l'anticipation, qui affecte plusieurs organes : le squelette, les muscles lisses, l'œil, le cœur, le système endocrinien et le système nerveux central.
| Dystrophie myotonique de Steinert | |
| Référence MIM | 160900 |
|---|---|
| Transmission | Dominante |
| Chromosome | 19 q13.2-q13.3 |
| Gène | DMPK |
| Empreinte parentale | Non |
| Mutation | Expansion de triplet |
| Mutation de novo | Rare |
| Nombre d'allèles pathologiques | Sans objet |
| Anticipation | Oui |
| Porteur sain | Sans objet |
| Prévalence | 1/20 000 |
| Maladie génétiquement liée | Aucune |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
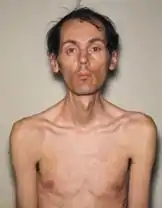
Les signes de cette maladie sont variés, allant d'une forme légère à une forme grave. Trois formes sont habituellement décrites selon l'âge d'apparition des premiers symptômes mais dont les limites ne sont pas toujours nettes : légère, classique et congénitale.
- La forme légère est caractérisée par une cataracte et une myotonie modérée. L'espérance de vie est normale.
- La forme classique est caractérisée par une faiblesse musculaire généralisée et une myotonie généralisée, une cataracte et des troubles de la conduction cardiaque. L'adulte peut perdre son autonomie et l'espérance de vie est réduite si le patient n'est pas suivi pour le cœur.
- La forme congénitale avec une hypotonie musculaire, souvent associée à une insuffisance respiratoire avec décès précoce. Le retard mental est fréquent dans cette forme.
La mutation est une expansion instable d'un triplet CTG du gène DMPK. Ce triplet est répété plus de 37 fois chez les personnes atteintes. Le nombre de répétitions du triplet CTG est généralement associé à la sévérité de la maladie ainsi qu'à l'âge d'apparition des symptômes.
Autres noms
- Myotonie dystrophique de type 1
- Dystrophie myotonique de type 1
Cause
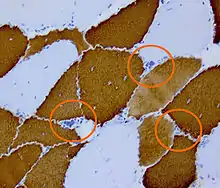
La maladie est provoquée par une mutation du gène DMPK (pour Dystrophy Myotonic Protein Kinase) situé sur le locus q13.32 du chromosome 19 codant la myotonine, une protéine kinase AMPc dépendante, dont le rôle précis est inconnu[1]. La mutation en cause est une expansion du triplet CTG dont le nombre dépasse 37 chez les personnes atteintes. Lorsque le nombre de répétition est supérieur à 50, la maladie se manifeste toujours. L'ARN issu du gène muté fixe la protéine MBNL1 qui n'est plus disponible pour ses autres actions[2].
Dans la forme congénitale, l'expansion du triplet atteint plusieurs milliers.
La genèse de la maladie fait intervenir le système ubiquitine-protéasome[3], une sur-expression du P53 ainsi qu'une inhibition du S6K1 et du mTOR[4] conduisant à une autophagie au niveau musculaire. Il également un épissage modifié du récepteur à l'insuline conduisant à une résistance à l'insuline[5].
Prévalence et incidence
Il s'agit de la cause la plus fréquente des dystrophies musculaires de l'adulte[6]. La prévalence de cette maladie est de 1 sur 100 000 au Japon et 1 sur 10 000 en Islande. La prévalence mondiale de cette maladie est de 1 sur 20 000. La prévalence mondiale la plus élevée atteint 189 sur 100 000 de population dans la région du Saguenay-Lac-Saint-Jean au Québec (Canada).
Description de la maladie
| Phénotype | Signes cliniques | Longueur du triplet | Âge du début des signes | Espérance de vie |
|---|---|---|---|---|
| Prémutation | Aucun | 35-50 | Sans objet | Sans objet |
| Moyenne | Cataracte Myotonie |
50-150 | 20 à 70 ans | 60 ans à normale si suivi cardiaque |
| Classique | Faiblesse musculaire Myotonie Cataracte Trouble de la conduction cardiaque Calvitie |
100-1000 | 10 à 30 ans | 48 ans à normale si suivi cardiaque et pulmonaire |
| Congénitale | Myotonie Détresse respiratoire Retard mental |
Supérieur à 2000 | 0 à 10 ans | 45 ans à normale si suivi cardiaque et pulmonaire |
Ce tableau est un tableau indicatif. Certains patients ne ressentent quasiment aucun symptôme à 500 répétitions alors que d'autres peuvent être gravement atteints à seulement 150. La rapidité d'évolution de la maladie peut différer d'un patient à un autre.
Diagnostic
Clinique
La présomption du diagnostic repose sur l'association de signes cliniques variés (par exemple cataracte, calvitie, troubles musculaires et cardiaques). La myotonie est le relâchement lent de la contraction musculaire volontaire (main qui reste serrée après une poignée de main, difficulté d'élocution due à la lenteur de décontraction de la langue par exemple).
Des signes d'atrophies musculaires peuvent aussi mettre sur la piste de la maladie :
- atrophie des muscles fléchisseurs du cou (souvent associés à un hyperdéveloppement des muscles sterno-cléido-mastoïdiens et des trapèzes par compensation musculaire) ;
- atrophie des 3es phalanges (orteils et doigts de la main).
Biologique
La certitude du diagnostic est obtenue par une technique de biologie moléculaire, avec un prélèvement sanguin. En France, le consentement de la personne prélevée est indispensable. La technique de TP-PCR est utilisée et suffit à éliminer les sujets sains (nombre de CTG inférieur à 37), hétérozygotes pour le nombre de répétitions du triplet CTG sur le gène DMPK (les deux allèles comportent moins de 37 répétitions). Chez les homozygotes sains et les hétérozygotes avec expansion (ils ont un allèle à moins de 37 répétitions, et un allèle à plus de 37, et généralement plus de 50 CTG), on réalise une vérification par Southern blot. Les hétérozygotes avec expansion sont atteints de la dystrophie myotonique de Steinert.
La présence d'un cas de dystrophie de Steinert au sein d'une famille doit déclencher une enquête familiale. Là encore, l'enquête génétique n'est possible qu'avec l'accord explicite des personnes prélevées en France.
Différentiel
- Myotonie dystrophique type 2 secondaire à une expansion de CCTG du gène ZNF9
- Myopathie à corps d'inclusion
- Myopathie avec surcharge en desmine
Complications
Une mort subite est possible, soit par troubles du rythme ventriculaire soit par trouble de la conduction cardiaque, et ce, d'autant qu'il existe des anomalies sur l'électrocardiogramme (PR supérieur à 240 millisecondes, bloc auriculo-venticulaire du 2e ou du 3e degré notamment)[7]. La pose d'un stimulateur cardiaque permet, dans ces cas, de limiter les risques de morts subites. La décision de cette pose se fait à la suite d'une exploration électrophysiologique du faisceau de HIS.
Le risque de survenue d'un cancer est augmenté[8].
Traitement
Il n'existe pas de traitement étiologique de cette maladie.
Traitement des symptômes
Il existe une grande variété de traitements qui visent à soulager les symptômes les plus fréquents de la maladie. Ces traitements ne sont pas forcément spécifiques de la maladie de Steinert. Leur remboursement est souvent pris en charge en totalité par l’assurance maladie.
Contre la myotonie, il faut éviter le froid et effectuer des exercices musculaires en ambiance chaude. Il est bon de faire des exercices physiques légers, fréquents mais peu intensifs. Des exercices de kinésithérapie douce peuvent être prescrits pour travailler sur la posture. La myotonie ne devrait justifier d’un traitement que si elle se révèle invalidante.
Deux médicaments existent pour contrer les symptômes, la méxilétine[9] et la quinidine. Ils sont rarement prescrits car peuvent poser des problèmes au niveau cardiaque (effet arythmogène).
Contre les troubles cardiaques, il est indispensable de suivre le patient régulièrement. Une visite cardiologique exhaustive chaque année (comprenant électrocardiogramme, Holter et échocardiographie) est primordiale. Il est envisageable de poser un pacemaker ou un défibrillateur si nécessaire pour suppléer une éventuelle défaillance cardiaque.
Contre les troubles de l’humeur et la somnolence, on peut traiter par le modafinil. En cas d’apnée du sommeil, on peut traiter par une prise en charge ventilatoire nocturne spécifique. Il est également possible d’envisager un traitement par antidépresseurs et un accompagnement psychologique.
Un traitement médicamenteux est adapté dans le cas de troubles endocriniens (notamment thyroïdiens). Un suivi de l’alimentation et un bilan lipidique sont indiqués pour lutter contre la résistance à l’insuline. Les médicaments hypo-choléstérolémiants sont en revanche contre-indiqués.
Recherche
Une piste de recherche est l'utilisation d'ARN antisens ciblant l'ARN issu de l'ADN muté[10].
D'autres pistes sont étudiées, notamment la molécule Isis–DMPK–2.5Rx [11], ainsi que l’usage d’un système enzymatique nommé CRISPR-CASP9 conçu pour couper les répétitions aberrantes CTG dans l’ADN des cellules Steinert[12]. Dans tous les cas, on se heurte aux problématiques classiques de la thérapie génique, à savoir introduire le « remède » dans l'ensemble des cellules du corps. Certaines molécules défixent le MBNL1, constituant une méthode potentielle d'amélioration de la maladie, sans intervention génétique[13]. Une douzaine de pistes sont actuellement en cours d'étude[14].
Notes et références
- (en) Brook JD, McCurrach ME, Harley HG et al. « Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member » Cell 1992;68:799-808
- Lin X, Miller JW, Mankodi A et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy, Hum Mol Genet, 2006;15:2087–2097
- Vignaud A, Ferry A, Huguet A et al. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin-proteasome pathway, Neuromuscul Disord, 2010;20:319–325
- Beffy P, Del Carratore R, Masini M, Furling D, Puymirat J, Masiello P, Simili M, Altered signal transduction pathways and induction of autophagy in human myotonic dystrophy type 1 myoblasts, Int J Biochem Cell Biol, 2010;42:1973–1983
- Savkur RS, Philips AV, Cooper TA, Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy, Nat Genet, 2001;29:40–47
- Machuca-Tzili L, Brook D, Hilton-Jones D, Clinical and molecular aspects of the myotonic dystrophies: a review, Muscle Nerve, 2005;32:1–18
- (en) Groh WJ, Groh MR, Saha C et al. « Electrocardiographic abnormalities and sudden death in myotonic dystrophy Type 1 » N Eng J Med. 2008;358:2688-97.
- (en) Gadalla SM, Lund M, Pfeiffer RM et al. « Cancer risk among patients with myotonic muscular dystrophy » JAMA 2011;306:2480-6.
- Logigian EL, Martens WB, Moxley RT IV et al. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1, Neurology, 2010;74:1441-1448
- Wheeler TM, Leger AJ, Pandey SK et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy, Nature, 2012;488:111-115
- « Pour la Science », N° 456, p. 24-32, octobre 2015
- Herrendorff R, Faleschini MT, Stiefvater A et al. Identification of plant-derived alkaloids with therapeutic potential for myotonic dystrophy type I, J Biol Chem, 2016;291:17165–17177
- « Le Portail d'Information sur la MALADIE DE STEINERT (DM 1) et la dystrophie myotonique proximale (Promm / DM 2) »
Liens externes
- Ressources relatives à la santé :
- GeneReviews
- Orphanet
- (en) Diseases Ontology
- (en) DiseasesDB
- (en + es) Genetic and Rare Diseases Information Center
- (en) Héritage mendélien chez l'humain
- (en) Héritage mendélien chez l'humain
- (en) Medical Subject Headings
- (en) NCI Thesaurus
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 160 900
- (en) Thomas D Bird, « Myotonic Dystrophy Type 1 » In : GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2005. .
- Article sur Orpha.net du Dr Alexandre Moerman et du Pr Sylvie Manouvrier
- Dystrophie myotonique de Steinert sur www.afm-telethon.fr
- Dystrophie myotonique de Steinert Québec Portail
 Portail de la médecine
Portail de la médecine  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire