Séquençage de l'ARN
Le séquençage de l'ARN (RNA-Seq, de l’anglais RNA sequencing), également appelé séquençage aléatoire du transcriptome entier (whole transcriptome shotgun sequencing en anglais)[1], est une technologie qui utilise le séquençage à haut débit (next-generation sequencing en anglais) pour identifier et quantifier l'ARN issu de la transcription du génome à un moment donné[2].
Introduction
Le transcriptome d'une cellule est dynamique, en constante évolution. Les technologies de séquençage à haut débit ont permis la lecture de l'ADN d'une cellule, base par base. Ces technologies permettent également la lecture de l'ARN dans une cellule, ce qui facilite la compréhension des mécanismes d'épissage alternatif de gènes, comprenant les modifications post-transcriptionnelles, les gènes de fusion, les polymorphismes nucléotidiques (SNP en anglais) et les changements dans l'expression génique[3]. En plus de l'ARNm, la technologie RNA-Seq permet d'identifier d'autres types d'ARN, tels que les microARN (miRNA en anglais), l'acide ribonucléique de transfert (ARNt, tRNA en anglais) et l'ARN ribosomal (ARNr, rRNA anglais)[4]. La technologie du RNA-Seq est utile pour déterminer les marges exon/intron des gènes et pour vérifier la localisation des extrémités 5' et 3' annotées antérieurement.
Les études réalisées avec la technologie RNA-Seq comprennent l'observation des changements qui se produisent dans une cellule au cours d'infections [5] et des changements dans l'expression génique pour les études portant sur le cancer[6].
Avant l'arrivée des technologies de séquençage à haut débit, les études de masse portant sur l'expression des gènes étaient principalement réalisées par le biais de la technologie des puces à ADN.
Méthodologie
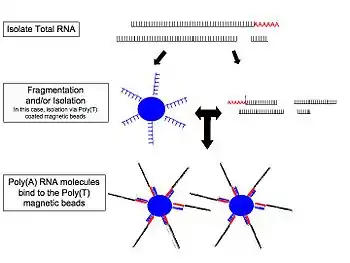
La méthodologie générale du RNA-Seq est décrite ci-contre. La méthode la plus courante est celle visant les ARN polyadénylés car ceux-ci représentent les ARNm stables et donc non ciblés pour dégradation, qui correspondent donc principalement à des ARNm dont le but est la traduction en protéine. De plus, la sélection des ARN polyadénylés permet l'élimination des ARN ribosomaux qui ne sont pas sujets à ce phénomène [7]. À noter qu'il existe d'autres protocoles ayant pour cible d'autres ARN, notamment les micro-ARN, ainsi que les ARN naissant ou encore ARN non codants. Enfin, bien que la méthode la plus courante vise l'ARN total d'une cellule, il existe certains protocoles visant à isoler les ARN nucléaires ou cytoplasmiques, selon le cadre de l'étude[8].
La création d'une librairie de séquençage peut varier de plateforme à plateforme de séquençage à haut débit[9], pour chacune desquelles il existe de nombreux kits dédiés à la création de différents types de librairies et pour l'adaptation des séquences résultantes aux exigences specifiques de leurs instruments. Cependant, dû à la nature des produits analysés, il existe des similarités entre chaque technologie, le principe général demeurant essentiellement le même. Fréquemment, lors d'analyses d'ARNm, la queue polyadénylée 3' (poly(A)) est ciblée de manière à assurer la séparation entre les ARN codants des ARN non-codants. Ceci est accompli par l'ajout covalent d'oligonucléoitides poly(T) au substrat. Actuellement, de nombreuses études utilisent des billes magnétiques à cet effet[10],[11],[12].
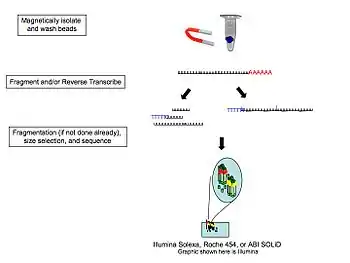
Des études portant sur des portions du transcriptome non polyadénylé ont montré que lors de l'utilisation de billes magnétiques poly(T), l'écoulement traversant peut contribuer à la découverte de gènes non-codants importants qui auraient autrement été manqués[10]. De plus, l'ARN ribosomal représentant plus de 90 % de l'ARN total d'une cellule donnée, des études ont montré que son élimination par le biais d'hybridation de sondes spécifiques visant ces ARN augmente très sensiblement la capacité d'un séquençage RNA-Seq à cibler le reste du transcriptome.
L'étape suivante est la transcription inverse (reverse transcription en anglais), qui vise à convertir les ARN en ADNc. Pour corriger le biais 5' de la transcription inverse provenant d'amorces aléatoires, et aussi bien pour réduire la formation de structures secondaires influençant les sites de liaison des amorces [11], une hydrolyse de l'ARN en fragments de 200-300 nucléotides est généralement utilisée de manière à résoudre ces problèmes potentiels. Cependant, il existe des limitations à cette méthode: même si de manière générale le corps des transcrits (correspondant au corps du gène) est converti efficacement en ADNc, il en va de moins pour les extrémités 5' et 3'. Les chercheurs choisiront donc d'appliquer ou d'ignorer cette étape selon les objectifs établis au cours d'une étude donnée.
Une fois l'ADNc synthétisé, celui-ci peut être fragmenté davantage de manière à atteindre la longueur de fragment désirée pour le système de séquençage utilisé, comme par exemple la technologie Illumina.
Le séquençage permet alors d'évaluer l'abondance des transcrits de chaque gène, c'est-à-dire le nombre de fois qu'un même transcrit a été séquencé. Ainsi, il est possible de déterminer l'expression relative des gènes dans une condition physiologique donnée, en comparaison à une condition physiologique de référence.
Notes et références
- Ryan D. Morin, Matthew Bainbridge, Anthony Fejes, Martin Hirst, Martin Krzywinski, Trevor J. Pugh, Helen McDonald, Richard Varhol, Steven J.M. Jones, and Marco A. Marra., « Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing », BioTechniques, vol. 45, no 1, , p. 81–94 (PMID 18611170, DOI 10.2144/000112900, lire en ligne)
- Chu Y, Corey DR, « RNA sequencing: platform selection, experimental design, and data interpretation », Nucleic Acid Ther, vol. 22, no 4, , p. 271–4 (PMID 22830413, PMCID 3426205, DOI 10.1089/nat.2012.0367)
- C. A. Maher, C. Kumar-Sinha, X. Cao et al., « Transcriptome sequencing to detect gene fusions in cancer », Nature, vol. 458, no 7234, , p. 97–101 (PMID 19136943, PMCID 2725402, DOI 10.1038/nature07638)
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS, « The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments », Nat Protoc, vol. 7, no 8, , p. 1534–50 (PMID 22836135, PMCID 3535016, DOI 10.1038/nprot.2012.086)
- F. Qian, L. Chung, W. Zheng et . etal, « Identification of Genes Critical for Resistance to Infection by West Nile Virus Using RNA-Seq Analysis », Viruses, vol. 5, no 7, , p. 1664–81 (PMID 23881275, DOI 10.3390/v5071664)
- J. Beane, J. Vick, F. Schembri et al., « Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq », Cancer Prev Res (Phila), vol. 4, no 6, , p. 803–17 (PMID 21636547, PMCID 3694393, DOI 10.1158/1940-6207.CAPR-11-0212)
- Shimyn Slomovic, David Laufer, Dan Geiger et Gadi Schuster, « Polyadenylation of ribosomal RNA in human cells », Nucleic Acids Research, vol. 34, , p. 2966–2975 (ISSN 1362-4962, PMID 16738135, PMCID 1474067, DOI 10.1093/nar/gkl357, lire en ligne, consulté le )
- Donald C. Rio, Manuel Ares, Gregory J. Hannon et Timothy W. Nilsen, « Preparation of cytoplasmic and nuclear RNA from tissue culture cells », Cold Spring Harbor Protocols, vol. 2010, , pdb.prot5441 (ISSN 1559-6095, PMID 20516179, DOI 10.1101/pdb.prot5441, lire en ligne, consulté le )
- Zhong Wang, Mark Gerstein et Michael Snyder, « RNA-Seq: a revolutionary tool for transcriptomics », Nature Reviews. Genetics, vol. 10, , p. 57–63 (ISSN 1471-0064, PMID 19015660, PMCID 2949280, DOI 10.1038/nrg2484, lire en ligne, consulté le )
- Ryan Morin, Matthew Bainbridge, Anthony Fejes et Martin Hirst, « Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing », BioTechniques, vol. 45, , p. 81–94 (ISSN 0736-6205, PMID 18611170, DOI 10.2144/000112900, lire en ligne, consulté le )
- Ali Mortazavi, Brian A. Williams, Kenneth McCue et Lorian Schaeffer, « Mapping and quantifying mammalian transcriptomes by RNA-Seq », Nature Methods, vol. 5, , p. 621–628 (ISSN 1548-7105, PMID 18516045, DOI 10.1038/nmeth.1226, lire en ligne, consulté le )
- « RNA Extraction/mRNA Isolation Protocols », sur www.protocol-online.org (consulté le )
Voir aussi
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire