Stavudine
La stavudine (2'-3'-didéhydro-2'-3'-didéoxythymidine ou d4T) est un médicament antirétroviral, c'est un inhibiteur nucléosidique de la transcriptase inverse (NRTI). Cette molécule est commercialisée sous le nom « Zerit ».
| Stavudine | |
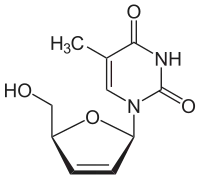
| |
| Structure chimique de la stavudine | |
| Identification | |
|---|---|
| Nom UICPA | D 2',3'-didéhydro-3'-déoxythymidine |
| Synonymes |
d4T |
| No CAS | |
| No ECHA | 100.169.180 |
| Code ATC | J05 |
| SMILES | |
| InChI | |
| Propriétés chimiques | |
| Formule | C10H12N2O4 [Isomères] |
| Masse molaire[1] | 224,213 3 ± 0,010 4 g/mol C 53,57 %, H 5,39 %, N 12,49 %, O 28,54 %, |
| Unités du SI et CNTP, sauf indication contraire. | |
Elle fait partie de la liste des médicaments essentiels de l'Organisation mondiale de la santé (liste mise à jour en )[2].
Historique
La stavudine ou D4T (2, 3’-didéhydro-2’, 3’ –didéoxythymidine) est un principe actif antiviral utilisé dans le traitement des personnes atteintes par le virus de l’immunodéficience humaine (VIH), qui provoque le syndrome d’immunodéficience acquise (SIDA).
Elle a été synthétisée pour la première fois en 1966 par le Dr Jerome Horwitz, de la Michigan Cancer Foundation pour son potentiel en tant qu'agent anticancéreux. À la suite de l'identification de l'activité anti-VIH de l’azidovudine (AZT) dans les années 1980, des activités de recherche ont eu pour objectif de découvrir des traitements optimaux de cette infection. Un certain nombre d'analogues nucléosidiques synthétisés précédemment ont donc été évalués. La stavudine (d4T) ayant une structure voisine de l'AZT, est donc apparue prometteuse pour le développement d'un traitement anti-VIH (mise en évidence des effets inhibiteurs de l'AZT et des didésoxynucléotides à la fin les années 1980). Hitchcock et Martin, deux chercheurs de l'Université de Yale, ont travaillé en étroite collaboration avec la fondation Prusoff pour caractériser cette variété d'analogues nucléosidiques, dont le D4T. L'Institut Rega synthétisant en parallèle le D4T, un accord de négociation a été signé pour des stratégies de demande de brevet.
L'AZT, les didésoxynucléotides ainsi que le D4T ayant déjà été décrits dans la littérature scientifique, la demande de brevet n'a pas été demandée pour sa composition chimique mais pour un procédé d'utilisation. Le brevet final a donc été accordé au groupe Prusoff et aux chercheurs de l’université de Yale en 1990. Ils ont mis en évidence l'activité antivirale de la stavudine dans des études cliniques chez l'homme et ont pu prouver son efficacité. Dans cette étude, ils ont comparé le D4T avec d’autres anti-VIH analogues didésoxynucléotides. Malgré sa toxicité, le D4T a été considéré comme étant un bon candidat pour un développement clinique. Ce constat s'est appuyé sur le fait que le D4T restait actif contre les souches résistantes à l'AZT du VIH, et présentait une toxicité moins importante que celui-ci. Ainsi, le D4T a été mis sur le marché en 1994 par l'industriel pharmaceutique Bristol-Myers Squibb (BMS) sous le nom de Zerit. En effet, l'Université de Yale a cédé les droits exclusifs d’exploitation sur son invention à cette firme en 1988 afin de commercialiser cette substance et pouvoir en produire des quantités industrielles.
Avec le temps, des effets secondaires ont été constatés à la suite de l'utilisation des analogues de désoxynucléosides responsables d'une inhibition de la fonction mitochondriale, de la lipodystrophie (dystrophie du tissu graisseux), de l'acidose lactique, et de la neuropathie périphérique. Des études de toxicité ont été menées concernant les didésoxynucléotides et ont permis de mettre en évidence la toxicité relativement importante de la stavudine.
En conséquence, seuls 7000 patients atteints du SIDA aux États-Unis sont toujours sous stavudine pour des raisons de sûreté (données IMS). Pourtant, son faible coût de fabrication a permis au D4T d'être une thérapie de première ligne dans le monde. En effet, il est fourni par les fabricants de génériques et est payé principalement à l'aide de fonds publics. Son faible coût a également permis l’accès aux traitements dans les pays en développement à partir de 2003 et a été le traitement de plus de 4 millions de personnes à la fin de l'année 2008. L'OMS et l'ONUSIDA estiment que près de 2 millions de patients sont sous un traitement à base de D4T encore aujourd’hui.
Synthèse

La stavudine ou D4T fut synthétisée pour la première fois par le scientifique américain Jerome Horwitz dans les années 1960 par rupture de la liaison entre le carbone entouré en vert et l'oxygène. L'alcool primaire correspondant, la stavudine, est ainsi formée.
Actuellement, il existe de nombreuses façons de synthétiser la stavudine. En effet, beaucoup de précurseurs du D4T peuvent servir à la synthétiser et il existe principalement deux procédés de réaction :
- un réarrangement structural pour obtenir le D4T ;
- retrait des groupements protecteurs de l'alcool primaire.
Réarrangement structural
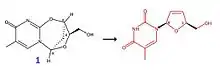
La molécule de formule brute C10H12N2O4 (correspondant au composé numéro 1) est un exemple de molécule pouvant servir à synthétiser du D4T en passant par un réarrangement structural. Cette molécule est d'abord mise en présence d'un sel d'imidazolium (à 50 °C), puis réagit avec NaH dans des conditions particulières (température de 100 °C, pendant 5 min), et forme ainsi la stavudine.
Notons que le rendement de cette réaction est de 93 %, alors que le rendement de la 1re synthèse de la stavudine était de 79 %.
Il existe d'autres molécules passant par ce même procédé, le mécanisme faisant intervenir le composé numéro 1 n'est qu'un exemple parmi d'autres.
Retrait des groupements protecteurs du OH
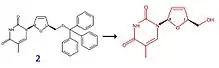
Parmi les différents procédés de synthèse de la stavudine, le plus courant et le plus utilisé consiste à déprotéger la fonction alcool primaire de la stavudine. Prenons l'exemple du composé 2, la fonction protectrice est un groupement triphénylméthyle. Ce réactif est mis en présence d'acide acétique dans de l'eau (tout ceci à une température de 50 °C pendant 2 h). On obtient finalement la stavudine avec un rendement de 93 %.
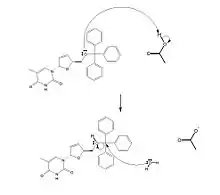
Mécanisme de dé-protection : l'acide acétique permet de protoner l'oxygène lié au groupement triphénylméthyle (cet oxygène se charge positivement), puis l'eau attaque l’atome de carbone central du groupement triphénylméthyle. Le triphénylméthanol est ainsi libéré.
Là encore, d'autres groupements peuvent substituer l'alcool primaire (par exemple, cela peut être un simple ester (COOCH3)), qui pourra être hydrolysé in fine.
Mécanisme d'action
La stavudine pénètre rapidement dans les cellules par diffusion non facilitée. Dans un grand nombre de cellules, la stavudine est phosphorylée de manière séquentielle en 5'-mono-, di- et triphosphate. La forme 5'-triphosphate est un inhibiteur de la transcriptase inverse, et a d’ailleurs un mécanisme comparable aux autres inhibiteurs de transcriptase inverse comme la zidovudine (AZT), ou l’entricitabine.
L’activation de la forme 5'-monophosphate par la cellule se fait grâce à une enzyme : la thymidine kinase. La stavudine monophosphate ne s'accumule pas dans la cellule. Constamment, il y aura des réactions transformant la forme monophosphate en diphosphate qui elle-même sera transformée en la forme triphosphate. Ainsi, la concentration intracellulaire de stavudine triphosphate augmente par rapport aux différentes concentrations extracellulaires de la molécule mère. Une fois formées, les stavudines triphosphates ont des demi-vies intracellulaires comprises entre 3 et 5h. La stavudine est un analogue de la thymidine dans lequel le groupe hydroxyle est remplacé par une double liaison entre deux atomes de carbone du cycle pentose. Comme les autres composés didésoxynucléosides, la stavudine-5'-triphosphate inhibe la réplication rétrovirale par compétition avec la molécule endogène : la désoxythymidine-triphosphate, servant à l’élongation de la molécule d’ADN.
La désoxythymidine-triphosphate est un substrat pour la transcriptase inverse du VIH. Cette enzyme virale permet de transcrire à l'envers, c’est-à-dire d'obtenir de l'ADN à partir d'ARN. Un virus ne peut pas se reproduire seul, il lui faut un hôte (une cellule), et c'est grâce à sa transcriptase inverse qu'il va fabriquer son propre ADN pour se reproduire, à partir des ARN qu'il trouvera dans la cellule qu'il parasite. Le principe est alors de bloquer l'élongation de la chaîne d'ADN du virus. Cela fonctionne par incorporation de la stavudine dans la formation de cet ADN viral. En effet, la molécule de stavudine va servir à bloquer la réplication de l'ADN, et donc le virus ne pourra plus proliférer. La molécule d’ADN est une succession de nucléotides qui sont reliés entre eux grâce à une liaison entre le 3'OH du ribose et le groupement phosphate du nucléotide suivant. Ce pont se fait sur l’hydroxyle du ribose, c’est ce qu’on appelle une liaison phosphodiester. Afin de se reproduire dans la cellule, le virus utilise les nucléotides de l'ADN pour transformer son ARN viral en ADN, et permettre ainsi sa prolifération. Or pour la stavudine, le groupement hydroxyle en 3' du ribose étant absent, il n'y a donc plus possibilité de polymérisation, ce qui empêche ainsi l’élongation de la molécule d’ADN, par conséquent le virus ne peut plus se servir de l'ADN pour se reproduire au sein de la cellule.
Autres caractéristiques
La résistance
En général, les patients qui montraient une résistance à la zidovudine, présentaient une résistance beaucoup moins importante pour la stavudine. Cependant, des études (réalisées en 1994) ont récemment montré, in vitro, qu’il existait des variants résistant à la stavudine. Lorsque ces variants étaient présents, ils présentaient une résistance 30 fois supérieure à la normale à la stavudine (provoquant une grande baisse de la CI50 de celle-ci). Cette résistance est due à une mutation du virus. En effet, au niveau du codon 50 (codant une thréonine), le virus a développé une mutagénèse spécifique lui attribuant une résistance face à la stavudine. Mais il existe également d’autres types de mutations qui rendent le virus du VIH résistant à d'autres traitements. En 1994, des chercheurs ont isolé une seconde souche mutante de VIH qui présentait une résistance 7 fois supérieure à la normale à la stavudine. Ce virus contient un changement de deux nucléotides au niveau du codon 75, ce qui provoque, entre autres, le remplacement d’une valine par une thréonine. Cette mutation confère une résistance croisée à la didanosine et à la zalcitabine (molécules pouvant faire partie de médicaments antirétroviraux). Notons que le virus résistant à la stavudine n'a pas pu être complètement isolé lors de ces études.
Toxicité
Plusieurs chercheurs ont comparé la cytotoxicité de la stavudine à celle de la zidovudine grâce à des tests, mais les résultats obtenus étaient différents. Dans certaines études, la stavudine était 20 à 100 fois moins toxique que la zidovudine, tandis que dans d'autres, la cytotoxicité des deux médicaments était similaire. Dans un test sur la toxicité neuronale in vitro, la stavudine a montré une toxicité moindre que la zidovudine. Par la suite, la didanosine et la zalcitabine ont été également présentées comme étant plus toxiques que la stavudine. La raison est que l'inhibition de la synthèse de l'ADN par la stavudine a lieu pour des concentrations inférieures par rapport à la zidovudine ou à la didanosine. Une diminution de la concentration de la molécule par rapport aux autres induit donc une diminution des effets secondaires de cette molécule, réduisant ainsi sa toxicité. L'interaction de la stavudine libre triphosphate et de la zidovudine libre triphosphate avec les ADN polymérases cellulaires (protéines servant à l’élongation de l'ADN) a été étudiée par plusieurs chercheurs. D'une manière générale, la stavudine triphosphate inhibe la γ-polymérase à des concentrations élevées. L'inhibition de la γ-polymérase a d’ailleurs été proposée comme un éventuel mécanisme de neuropathie périphérique induite par le médicament chez les patients infectés par le VIH.
Traitement
Posologie
Zerit contient comme excipients à effet notoire du lactose monohydrate et du lactose anhydre. Ce médicament est classé comme : -médicament à prescription restreinte -médicament à prescription initiale hospitalière -médicament à surveillance particulière.
La stavudine est administrée deux fois par jour par voie orale souvent sous la forme de gélules ou de poudre pour suspension buvable. La dose recommandée est de : - 40 mg toutes les 12 heures pour les patients adultes de 60 kg - 30 mg toutes les 12 heures pour les patients adultes pesant moins de 60 kg - 1 mg/kg toutes les 12 heures pour les enfants âgés de plus de 3 ans et pesant moins de 30 kg.
La dose doit être réduite pour les patients souffrant d’insuffisance rénale (il faut un suivi du patient pour vérifier la fonction rénale pendant la durée de traitement).
Les effets secondaires
* Les effets secondaires rares :
- effet sur le pancréas (pancréatite = inflammation du pancréas) qui se manifeste par une douleur abdominale intense avec ou sans nausées et vomissements - acidose lactique qui peut être mortelle : accumulation d'acide lactique dans le sang - troubles osseux pouvant apparaître.
* Les effets secondaires à long terme :
- Lipoatrophie : diminution de la quantité de tissus graisseux au niveau de la peau et redistribution des graisses corporelles. Il existe des traitements comme le sculptra et le bio-alcamid (ils agissent en stimulant la production de collagène) ; de plus, l’hormone de croissance humaine et les stéroïdes anabolisants sont d’autres traitements possibles. - Des neuropathies périphériques : maladie des nerfs périphériques qui se caractérise le plus souvent par un engourdissement persistant, des fourmillements ou des douleurs avec des sensations de brûlure et de picotement au niveau des pieds ou des mains. - De rares cas de leucopénie ont été observés (diminution du nombre de globules blancs pouvant augmenter le risque d’infection). - Effet sur le foie (hépatite). - Troubles rénaux : calculs rénaux. - Syndrome de reconstitution immunitaire : le système immunitaire peut se renforcer lors de la prise de stavudine, ainsi le système immunitaire du patient peut combattre des maladies et infections qui étaient au stade latent dans l’organisme. - L'hyperlipédimie, qui se traduit par une élévation du taux de triglycérides dans le sang (des prises de sang régulières au cours du traitement sont nécessaires pour le patient).
* Les effets secondaires fréquents :
-fatigue -troubles du sommeil, insomnies -perte de poids -maux de tête -nausées -diarrhées -des douleurs articulaires et musculaires -troubles psychiatriques : dépression.
Les allergies et l'alcool
Des réactions allergiques peuvent survenir sous forme de problèmes de peau (rougeurs, urticaires) ou de problèmes respiratoires. De plus, Zerit contient du sucre de lait (lactose), ainsi les personnes intolérantes au lactose et sous stavudine peuvent souffrir de nausées et de diarrhées. Il faut également noter qu'une consommation d'alcool sous stavudine peut augmenter le risque de problème de foie et de pancréas et donc aggraver les effets secondaires.
Grossesse et allaitement
On ne connaît pas les effets de la stavudine chez la femme enceinte et on ignore encore les effets nocifs sur le développement du fœtus. Cependant, des anomalies congénitales et des avortements ont été notés. L’allaitement n'est pas recommandé, voire fortement déconseillé pour les femmes atteintes par le VIH, puisque le risque de contaminer le bébé par le lait maternel n'est pas nul.
Trithérapie
Généralement, trois antiviraux sont prescrits simultanément dans le traitement de l'infection par le VIH afin d'éviter que le virus résiste à ces principes actifs. C'était le cas pour le D4T qui fut commercialisé avec la lamivudine (inhibiteur nucléosidique de la transcriptase inverse) et le névirapine (inhibiteur non nucléosidique de la transcriptase inverse) sous le nom générique « Triomune » dans les pays en développement. Cependant, ce générique n'est plus utilisé dans la trithérapie prescrite au début de la maladie pour cause d'effets indésirables importants.
Interactions médicamenteuses
Des interactions entre la stavudine et d'autres médicaments tels que la didanosine, la doxorubicine, l'hydroxyurée, la ribavirine ou la zidovudine peuvent avoir lieu, entraînant une aggravation des effets secondaires dans certains cas, ou l'inhibition de l'activité antivirale du D4T dans d'autres cas. Par exemple pour des hautes concentrations, la zidovudine sous sa forme monophosphate empêche la phosphorylation du D4T en sa forme active triphosphate. En effet, la zidovudine comme la stavudine est un substrat de l'enzyme thymidine kinase et donc se fixe sur les récepteurs de celle-ci . La zidovudine ayant une meilleure affinité envers les récepteurs de la thymidine kinase, elle s'y fixe. Les récepteurs sont donc saturés et la stavudine ne peut plus s'y fixer. La zidovudine est donc un antagoniste du D4T. Autre exemple, la stavudine associée avec la didanosine entraîne une augmentation de la toxicité mitochondriale.
Notes et références
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- (en) WHO Model List of Essential Medicines, 18th list, avril 2013
Bibliographie
Early nucleoside reverse transcriptase inhibitors for the treatment of HIV: A brief history of stavudine (D4T) and its comparison with other dideoxynucleosides ; 2010, John C. Martin, Michael J.M. Hitchcock, Erik De Clercq, William H. Prusoff;
Liens externes
- Page spécifique dans la base de données sur les produits pharmaceutiques (Canada)
- Compendium suisse des médicaments : spécialités contenant Stavudine
- Page spécifique sur le Répertoire Commenté des Médicaments, par le Centre belge d'information pharmacothérapeutique
- Page spécifique sur le Vidal.fr
- acticle sur l'historique du D4T
- Réactions répertoriées sur scifinder
- les trithérapies dans le traitement du VIH
- article sur les effets secondaires
- dossier sur les interactions médicamenteuses
 Portail de la médecine
Portail de la médecine  Portail de la virologie
Portail de la virologie  Portail de la pharmacie
Portail de la pharmacie  Portail de la chimie
Portail de la chimie