Syndrome d'Ehlers-Danlos
Les syndromes d'Ehlers-Danlos (SED), ou EDS (en anglais, pour Ehlers-Danlos syndrome) regroupent des affections toutes d'ordre génétique, rares ou orphelines, nommées d'après les recherches du Danois Edvard Ehlers (en) et du Français Henri-Alexandre Danlos au tout début du XXe siècle.
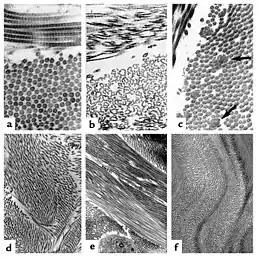
(a) Fibrilles de collagène normales, régulièrement disposées et uniformes en taille.
(b) les fibrilles chez un patient avec un SED de type dermatosparaxis montrent une altération importante de la morphologie des fibrilles avec des conséquences importantes sur la force de tension des tissus conjonctifs.
(c) Les patients avec un SED classique montrent des fibrilles composites.
(d) Chez un patient avec une anomalie du gène TNX les fibrilles sont uniformes en taille et il n'y a pas de fibrille composite.
(e) En cas d'absence totale d'expression du gène TNX les fibrilles sont moins denses et mal alignées.
| Symptômes | Hyperélasticité cutanée (d) et hypermobilité articulaire (d) |
|---|
| Spécialité | Génétique médicale |
|---|
| CISP-2 | L82 |
|---|---|
| CIM-10 | Q79.6 (ILDS Q82.817) |
| CIM-9 | 756.83 |
| DiseasesDB | 4131 |
| MedlinePlus | 001468 |
| eMedicine |
1114004 ped/654 |
| MeSH | D004535 |
| Patient UK | Ehlers-danlos-syndrome-pro |
![]() Mise en garde médicale
Mise en garde médicale
Maladie génétique par atteinte du collagène (élastorrhexie), les SED sont systémiques et liés à une anomalie du tissu conjonctif qui constitue 80 % du corps humain. La symptomatologie est alors si riche et polymorphe qu'elle complexifie la pose du diagnostic et renforce les errances du corps médical, dérouté devant autant de symptômes. Le point commun à toutes ces affections est l'hyperlaxité articulaire, l'hyperélasticité cutanée et la fragilité des tissus conjonctifs[1].
Il est important de se reporter aux articles spécifiques propres à chaque forme particulière citées ci-dessous, en particulier des deux premières.
Classification
Proposition de simplification du classement de 1988
En 1997, un groupe de chercheurs a proposé de réduire à six types la classification des SED[2] :
- syndrome d'Ehlers-Danlos type hypermobile (type III). L'apparition statistique est au centre des débats entre chercheurs ; elle serait de 1 personne sur 20 000. Il y a environ 3 000 patients diagnostiqués en France sur 66 millions de Français ;
- syndrome d'Ehlers-Danlos type classique (ancien type I/II). Même remarque que pour le SED hypermobile. La prévalence hypothétique serait de 1 sur 20 000. Il y a environ 4 000 patients diagnostiqués en France. Les chiffres de la prévalence sont remis en cause et récemment revus à la baisse dans les derniers travaux de recherche car beaucoup trop forte par rapport au nombre de cas officiellement diagnostiqués ;
- syndrome d'Ehlers-Danlos type vasculaire (type IV). La prévalence serait de 1/150000. Officiellement : 500 cas répertoriés en France ;
- syndrome d'Ehlers-Danlos type oculo-cypho-scoliotique (type VI). Environ une quarantaine de cas connus dans le monde ;
- syndrome d'Ehlers-Danlos type arthro-chalasique (type VII). Moins d'une trentaine de cas connus dans le monde ;
- syndrome d'Ehlers-Danlos type dermato-sphixique (type VIII). Une dizaine de cas connus dans le monde.
L'existence d'autres types que les trois premiers fait débat au sein des spécialistes ; certains pensent comme le Professeur Claude Hamonet que les types VI /VII et VIII n'existeraient pas et ne seraient que des manifestations et expressions diverses du type III. L'équipe de recherche Belgique/France/Canada irait jusqu'à en dénombrer aujourd'hui une douzaine avec des formes jadis ignorées et encourage les patients ayant un type III très sévère à faire des tests génétiques car il pourrait s'agir pour certains d'une autre forme.
Des syndromes apparentés sont liés à l'atteinte de gènes codant d'autres constituants de la matrice comme les protéoglycanes, à transmission autosomale récessive[3]. Le type vasculaire est le plus grave, avec un pronostic vital pessimiste. Pour les autres, la sévérité peut considérablement varier d'un patient à l'autre, comme dans le type III, considéré comme lourd lorsque l'apparition d'un cancer ou d'un syndrome parkinsonien vient bouleverser la vie du patient.
Classification internationale de 2017
En 2017, de nouvelles données génétiques et cliniques ont amené la communauté internationale a adopter une nouvelle classification, comportant en sus des types déjà cités, les autres types suivants :
- syndrome d’Ehlers-Danlos de type classic-like (SEDcl), proche du Syndrome d'Ehlers-Danlos type classique, mais à transmission autosomique récessive, dû à un déficit en ténascine X ;
- syndrome d’Ehlers-Danlos de type spondylodysplasique (SEDsp), à transmission autosomique récessive, dû à une mutation de l'un des gènes B4GALT7, B3GALT6 ou SLC39A13, et présentant un tableau différent en fonction du gène concerné, avec en tableau commun une petite taille, une incurvation des membres, une hypotonie musculaire et plus rarement un retard psychomoteur ;
- syndrome d’Ehlers-Danlos de type « musculocontractural » (SED mc), à transmission autosomique récessive, souvent dû à des mutations du gène CHST14, et plus exceptionnellement du gène DSE (contractures congénitales en adduction-flexion, pieds équinovarus, luxations récurrentes, déformation thoracique, pneumothorax, fragilité cutanée avec cicatrices atrophiques et troubles ophtalmologiques) ;
- Brittle Cornea Syndrome (BCS), de transmission autosomique récessive, due à des mutations des gènes ZNF469 et PRDM5. Le tableau comporte des signes ophtalmologiques sévères, une surdité et des troubles musculosquelettiques ;
- syndrome d’Ehlers-Danlos de type myopathique (SEDm), dû à des mutations du gène COL12A1, avec hypotonie musculaire congénitale, contractures articulaires proximales, hyperlaxité des articulations distales et retard de développement moteur ;
- syndrome d’Ehlers-Danlos de type parodontal (SEDp), de transmission autosomique dominante, dû à des mutations des gènes C1R ou C1S. À l'origine d'une périodontopathie précoce sévère, il prédispose aux infections, et il est caractérisé par des plaques d’hématomes prétibiaux ;
- syndrome d’Ehlers-Danlos de type cardiaque valvulaire (SEDcv), de transmission autosomique récessive, dû à des mutations du gène COL1A2 et responsable d’une valvulopathie sévère progressive[1].
Notes et références
- Centre de référence des Maladies Osseuses Constitutionnelles, Sites constitutifs pour les Syndromes d'Ehlers-Danlos Non Vasculaires, Filière de santé maladies rares de l’os, du calcium et du cartilage (OSCAR), , 211 p. (lire en ligne)
- (en) Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ, « Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) », Am J Med Genet, vol. 77, no 1, , p. 31–7. (PMID 9557891, DOI 10.1002/(SICI)1096-8628(19980428)77:1<31::AID-AJMG8>3.0.CO;2-O)
- (en) Malfait F, Kariminejad A, Van Damme T, Gauche C, Syx D, Merhi-Soussi F, Gulberti S, Symoens S, Vanhauwaert S, Willaert A, Bozorgmehr B, Kariminejad MH, Ebrahimiadib N, Hausser I, Huysseune A, Fournel-Gigleux S, De Paepe A, « Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder », Am J Med Genet, vol. 92, no 6, , p. 935-45. (PMID 23664118, PMCID PMC3675258, DOI 10.1016/j.ajhg.2013.04.016)
Liens externes
- Fiche d'information sur le syndrome d'Ehlers-Danlos classique sur Orphanet
- Publication Internationale Ehlers-Danlos 2017 [PDF] (4,9 Mo) sur UNSED.org
 Portail de la médecine
Portail de la médecine