Syndrome de Brugada
Le syndrome de Brugada est une maladie génétique rare caractérisée par un sus-décalage du segment ST (en) au niveau des dérivations précordiales droites V1, V2 et V3, et un aspect de bloc de branche droit à l'électrocardiogramme associés à un risque élevé d'arythmie ventriculaire pouvant entraîner syncope et mort subite, sur un cœur structurellement sain. La transmission se fait sur un mode autosomique dominant et la pénétrance est variable. Des mutations génétiques entraînent des anomalies au niveau des canaux ioniques. L'âge moyen du premier épisode clinique est de 40 ans, avec une forte prédominance masculine. La prévalence estimée est d'environ 1/1000 dans les pays asiatiques, probablement plus faible ailleurs. Le pronostic est grave chez les patients présentant des symptômes et la mort subite peut être prévenue par la pose d'un défibrillateur automatique implantable.
Pour les articles homonymes, voir Brugada.
| Spécialité | Cardiologie |
|---|
| CIM-10 | + I45.130 Q24.8 + I45.130 |
|---|---|
| CIM-9 | 746.89 |
| OMIM | 601144 |
| DiseasesDB | 31999 |
| eMedicine | 163751 |
| MeSH | D053840 |
![]() Mise en garde médicale
Mise en garde médicale
Historique
Ce syndrome a été décrit pour la première fois en 1992 par les frères Pedro et Josep Brugada[1].
Étiologie
Le syndrome de Brugada est une canalopathie sodique.
L'une des causes est une mutation du gène SCN5A situé sur le chromosome 3[2]. Près de 300 mutations différentes ont été identifiées sur ce gène mais ces mutations ne sont retrouvées que dans un cas de Brugada sur cinq[3].
Il s'agit d'une maladie de transmission autosomique dominante avec une pénétrance faible (peu de porteurs de la mutation présentent les signes de la maladie).
Les gènes SCN10A, HEY2[4], GPDL1 (le gène de l'enzyme glycerol-3 phosphate dehydrogenase-1 like)[5] ou CACNA1C[6] peuvent être en cause. Pour les autres patients atteints sans avoir de mutation du SCN5A, la maladie est bien là, mais aucun gène responsable n'est identifié dans trois quarts des cas[3]. L'hypothèse d'une origine multifactorielle est évoquée[7]. Le typage génétique ne semble pas avoir jusque-là un intérêt pronostique[8]. Cela semble moins vrai avec les avancées de la recherche[9].
Au niveau électrophysiologique, il semble exister une hétérogénéité des délais de conduction dans le ventricule droit qui pourrait être le substrat au déclenchement et la pérennisation des troubles rythmiques[10]. Elle serait associée à une conduction ralentie au niveau de la chambre de chasse du ventricule droit[11] et à des bas voltages[12], avec fibrose et un déficit en jonctions communicantes à cet endroit[13]. Un mécanisme inflammatoire à ce niveau est souvent associé[12].
Épidémiologie
La fréquence est estimée à 1 sur 1 000 dans les populations asiatiques où le syndrome de mort brutale au cours du sommeil est fréquent ; il s'agit de la seconde cause de mort subite de l'homme jeune en Asie, après les accidents de la route.
L'âge moyen du diagnostic ou de la mort subite est de 40 ans plus ou moins 22 ans, avec des extrêmes allant entre deux jours de vie jusqu'à plus de 80 ans.
Il semble plus fréquent chez les hommes, ces derniers ayant des formes plus graves[14].
Description
Le diagnostic de syndrome de Brugada repose sur l'association d'anomalies à l'ECG (sus-décalage du segment ST), et d'événements de type syncope ou mort subite. Dans un nombre de cas croissant, les cardiologues sont amenés à se prononcer sur la réalité de ce diagnostic chez des patients asymptomatiques présentant uniquement un aspect anormal à l'ECG (sus-décalage du segment ST).
En pratique, il faut distinguer le syndrome de Brugada diagnostiqué d'un simple aspect de Brugada sur l'électrocardiogramme, souvent découvert fortuitement, et dont les risques sont très variables (mais en moyenne inférieurs).
Diagnostic
Les éléments intervenant dans le diagnostic sont :
- antécédents familiaux de syndrome de Brugada, de mort subite ;
- antécédents personnels de mort subite ressuscitée ;
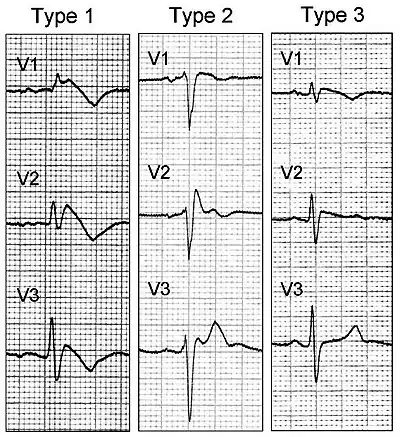
- à l'électrocardiogramme : aspect de bloc de branche droit associé à un sus-décalage du segment ST et des anomalies de l'onde T dans les dérivations précordiales droites (V1 à V3). On décrit trois types d'anomalies ECG :
- le type 1 : sus-décalage du segment ST en forme de dôme,
- le type 2 : sus-décalage du segment ST en forme de selle de cheval,
- le type 3 : même figure sur l'électrocardiogramme que le type 2 mais en aplati,
- positivité du test à l'ajmaline ou à la Flécaïne[15] ;
- identification de la mutation du gène codant SCN5A.
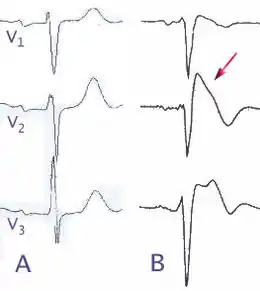
La distinction en trois types suivant l'électrocardiogramme est parfois délicate, car ce dernier peut varier d'un jour à l'autre chez le même individu, passant d'un type à l'autre[16]. Le sus-décalage peut augmenter à la phase de récupération lors d'une épreuve d'effort dans un tiers des cas, et serait alors péjoratif en termes de pronostic[17]. De plus, le type 1 est retrouvé plus fréquemment si les électrodes sont placées sur la poitrine un espace intercostal plus haut que d'habitude[18]. D'autres anomalies électriques peuvent être associées : ainsi, une repolarisation précoce (sus décalage concave le haut du segment ST) dans les territoires inférieurs se voit dans un cas sur 10 et serait un critère péjoratif[19].
Prise en charge
Des recommandations sur la prise en charge du Brugada ont été publiées en 2005[20]. D'autres, américaines et européennes, concernant les troubles du rythme d'origine génétique, ont été publiées en 2013[21].
Dans tous les cas, il faut éviter ou traiter toute circonstance déclenchante connue d'un Brugada de type I. Il convient, en particulier, de traiter rapidement toute fièvre[8].
La prise en charge dépend du type d'anomalie ECG, du résultat du test pharmacologique, des antécédents familiaux de mort subite, des symptômes et troubles du rythme présentés par le patient. Dans le cas d'un syndrome de Brugada complet et typique avec antécédent personnel d’arrêt cardiaque récupéré et d’absence de doute diagnostique, l'indication de la pose d'un défibrillateur automatique implantable (DAI) est communément admise. Cette indication est plus discutée dans les autres cas (notamment anomalie isolée de l'électrocardiogramme), le risque de mort subite étant notablement plus faible. Il n'est cependant pas nul et le problème repose essentiellement sur la stratification de ce risque.
Il existe au début des années 2000 un débat scientifique qui n'est toujours pas tranché sur la conduite à tenir en cas d'aspect électrocardiographique spontané de type 1 chez un patient asymptomatique. Une exploration électrophysiologique était précédemment recommandée avec protocole de stimulation ventriculaire droite programmée afin de rechercher un trouble du rythme ventriculaire déclenché, les premières données semblant montrer qu'il s'agissait d'un critère péjoratif[22] mais cela n'a pas toujours été confirmé par des données plus récentes[23] et son intérêt reste discuté[24]. De même, un antécédent familial de mort subite ne semble pas augmenter ce risque chez la personne asymptomatique[23]. La « sanction » thérapeutique varie, en fonction de la balance entre bénéfices et risques, d'une absence de recommandation particulière (sujet dont on estime que le risque personnel est celui de la population générale saine), jusqu'à la mise en place d'un défibrillateur automatique implantable qui assurera un choc électrique (défibrillation) en cas de survenue de troubles du rythme ventriculaire.
Il n'existe pas de traitement médicamenteux prévenant les troubles du rythme ventriculaire dans le syndrome de Brugada. La quinidine est employée par certains, pour l'instant de manière empirique[25].
L'ablation par radiofréquence, par voie épicardique d'une zone située au niveau de la chambre de chasse du ventricule droit, permet la disparition de l'anomalie de l'ECG[26] mais son utilité quant à la prévention de la mort subite reste à établir[27].
Conseil génétique
Les données actuelles sur le mécanisme de cette maladie étant en faveur d'un grand nombre de cas de mutations sur les récepteurs de canaux notamment sodiques au sein des cellules myocardiques, il est parfois possible de tenter la recherche de mutations familiales lorsqu'existent des arguments pour de multiples cas familiaux. Cette recherche est cependant discutable : en pratique, la présence d'une mutation ne semblant pas être corrélée avec une augmentation du risque de survenue d’évènements graves[23].
Notes et références
- (en) Brugada P, Brugada J, « Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report », J Am Coll Cardiol, vol. 20, no 6, , p. 1391-6. (PMID 1309182, DOI 10.1016/0735-1097(92)90253-j, résumé)
- (en) Chen Q, Kirsch GE, Zhang D, Brugada R, Wang Q et al., « Genetic basis and molecular mechanism for idiopathic ventricular fibrillation », Nature, vol. 392, no 6673, , p. 293–6. (PMID 9521325, DOI 10.1038/32675, résumé)
- (en) Kapplinger JD, Tester DJ, Alders M, Benito B, Ackerman MJ et al., « An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing », Heart Rhythm, vol. 7, no 1, , p. 33–46. (PMID 20129283, PMCID PMC2822446, DOI 10.1016/j.hrthm.2009.09.069, lire en ligne [html])
- (en) Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Redon R et al., « Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death », Nat Genet, vol. 45, no 9, , p. 1044–9. (PMID 23872634, PMCID PMC3869788, DOI 10.1038/ng.2712, lire en ligne [html])
- (en) London B, Michalec M, Mehdi H, Zhu X, Dudley Jr SC et al., « Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias », Circulation, vol. 116, no 20, , p. 2260–8. (PMID 17967977, PMCID PMC3150966, DOI 10.1161/CIRCULATIONAHA.107.703330, résumé)
- (en) Novelli V, Memmi M, Malovini A, Mazzanti A, Priori SG et al., « Role of CACNA1C variants in Brugada syndrome: clinical aspects and genetic testing strategies », Eur Heart J, vol. 41, no Supplement_2, , ehaa946.3584 (DOI 10.1093/ehjci/ehaa946.3584, lire en ligne [html])
- (en) Probst V, Wilde AAM, Barc J, Sacher F, Schott J-J et al., « SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome », Circ Cardiovasc Genet, vol. 2, no 6, , p. 552–7. (PMID 20031634, DOI 10.1161/CIRCGENETICS.109.853374, lire en ligne [html])
- (en) Brugada J, Campuzano O, Arbelo E, Sarquella-Brugada G, Brugada R, « Present status of Brugada syndrome: JACC state-of-the-art review », J Am Coll Cardiol, vol. 72, no 9, , p. 1046-59. (PMID 30139433, DOI 10.1016/j.jacc.2018.06.037)
- (en) Postema PG, Walsh R, Bezzina CR, « Illuminating the path from genetics to clinical outcome in Brugada syndrome », Eur Heart J, vol. 42, no 11, , p. 1091–3. (PMID 33444429, PMCID PMC7955964, DOI 10.1093/eurheartj/ehaa994, lire en ligne [html])
- (en) Lambiase PD, Ahmed AK, Ciaccio EJ et al., « High-density substrate mapping in Brugada syndrome: combined role of conduction and repolarization heterogeneities in arrhythmogenesis », Circulation, vol. 120, no 2, , p. 106-17, 1-4. (PMID 19564561, DOI 10.1161/CIRCULATIONAHA.108.771401, résumé)
- (en) Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJG, de Bakker JMT et al., « Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic and computational study », Circulation, vol. 112, no 18, , p. 2269–77. (PMID 16267250, DOI 10.1161/CIRCULATIONAHA.105.532614, lire en ligne)
- (en) Pieroni M, Notarstefano P, Oliva A et al., « Electroanatomic and pathologic right ventricular outflow tract abnormalities in patients with Brugada syndrome », J Am Coll Cardiol, vol. 72, no 22, , p. 2747-57. (PMID 30497561, DOI 10.1016/j.jacc.2018.09.037, lire en ligne [html])
- (en) Nademanee K, Raju H, de Noronha SV, Papadakis M, Behr ER et al., « Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome », J Am Coll Cardiol, vol. 66, no 18, , p. 1976-86. (PMID 26516000, PMCID PMC4631798, DOI 10.1016/j.jacc.2015.08.862, lire en ligne [html])
- (en) Benito B, Sarkozy A, Mont L, Henkens S, Brugada J et al., « Gender Differences in Clinical Manifestations of Brugada Syndrome », J Am Coll Cardiol, vol. 52, no 19, , p. 1567-73. (PMID 19007594, DOI 10.1016/j.jacc.2008.07.052, résumé)
- (en) Brugada R, Brugada J, Antzelevitch C et al., « Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts », Circulation, vol. 101, no 5, , p. 510–5. (PMID 10662748, DOI 10.1161/01.cir.101.5.510, lire en ligne [html])
- (en) Richter S, Sarkozy A, Veltmann C, Chierchia G-B, Brugada P et al., « Variability of the diagnostic ECG pattern in an ICD patient population with Brugada syndrome », J Cardiovasc Electrophysiol, vol. 20, no 1, , p. 69–75. (PMID 18775043, DOI 10.1111/j.1540-8167.2008.01282.x, résumé)
- (en) Makimoto H, Nakagawa E, Takaki H, Yamada Y, Shimizu W et al., « Augmented ST-segment elevation during recovery from exercise predicts cardiac events in patients with Brugada syndrome », J Am Coll Cardiol, vol. 56, no 19, , p. 1576–84. (PMID 21029874, DOI 10.1016/j.jacc.2010.06.033, résumé)
- (en) Shimizu W, Matsuo K, Takagi M et al., « Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve-lead electrocardiograms », J Cardiovasc Electrophysiol, vol. 11, no 4, , p. 396–404. (PMID 10809492, DOI 10.1111/j.1540-8167.2000.tb00334.x, résumé)
- (en) Sarkozy A, Chierchia GB, Paparella G et al., « Inferior and lateral electrocardiographic repolarization abnormalities in Brugada syndrome », Circ Arrhythm Electrophysiol, vol. 2, no 2, , p. 154–61. (PMID 19808460, DOI 10.1161/CIRCEP.108.795153, lire en ligne)
- (en) Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Wilde A et al., « Brugada syndrome: report of the second consensus conference », Heart Rhythm, vol. 2, no 4, , p. 429–40. (PMID 15898165, DOI 10.1016/j.hrthm.2005.01.005)
- (en) SG Priori, AA Wilde, M Horie, Cho Y, Tracy C et al., « HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013 », Heart Rhythm, vol. 10, no 12, , p. 1932-63. (PMID 24011539, DOI 10.1016/j.hrthm.2013.05.014, lire en ligne [html])
- (en) Brugada P, Brugada R, Mont L, Rivero M, Brugada J et al., « Natural history of Brugada syndrome: the prognostic value of programmed electrical stimulation of the heart », J Cardiovasc Electrophysiol, vol. 14, no 5, , p. 455–57. (PMID 12776858, DOI 10.1046/j.1540-8167.2003.02517.x, résumé)
- (en) Probst V, Veltmann C, Eckardt L, Meregalli PG, Wilde AAM et al., « Long-term prognosis of patients diagnosed with Brugada syndrome: results from the Finger Brugada Syndrome Registry », Circulation, vol. 121, no 5, , p. 635-43. (PMID 20100972, DOI 10.1161/CIRCULATIONAHA.109.887026, résumé)
- (en) Sroubek J, Probst V, Mazzanti A, Delise P, Lubitz SA et al., « Programmed ventricular stimulation for risk stratification in the Brugada syndrome: a pooled analysis », Circulation, vol. 133, no 7, , p. 622-30. (PMID 26797467, PMCID PMC4758872, DOI 10.1161/CIRCULATIONAHA.115.017885, lire en ligne [html])
- (en) Viskin S, Wilde AAM, Tan HL, Antzelevitch C, Belhassen B et al., « Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry », Heart Rhythm, vol. 6, no 3, , p. 401–4. (PMID 19251219, PMCID PMC3005559, DOI 10.1016/j.hrthm.2008.11.030, lire en ligne [html])
- (en) Pappone C, Brugada J, Vicedomini G, Ciconte G, Santinelli V et al., « Electrical substrate elimination in 135 consecutive patients with Brugada syndrome », Circ Arrhythm Electrophysiol, vol. 10, no 5, , e005053. (PMID 28500178, DOI 10.1161/CIRCEP.117.005053, lire en ligne [html])
- (en) Viskin S, « Radiofrequency ablation of asymptomatic Brugada syndrome: don't go burning my heart », Circulation, vol. 137, no 18, , p. 1883-4. (PMID 29530885, DOI 10.1161/CIRCULATIONAHA.117.032624, lire en ligne [html])
Annexes
Association de patients
Articles connexes
Bibliographie
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:601144
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 Les auteurs de cet article sont les docteurs Brugada.
 Portail de la médecine
Portail de la médecine