DNA Sequencing Techniques
While techniques to sequence proteins have been around since the 1950s, techniques to sequence DNA were not developed until the mid-1970s, when two distinct sequencing methods were developed almost simultaneously, one by Walter Gilbert's group at Harvard University, the other by Frederick Sanger's group at Cambridge University. However, until the 1990s, the sequencing of DNA was a relatively expensive and long process. Using radiolabeled nucleotides also compounded the problem through safety concerns. With currently-available technology and automated machines, the process is cheaper, safer, and can be completed in a matter of hours. The Sanger sequencing method was used for the human genome sequencing project, which was finished its sequencing phase in 2003, but today both it and the Gilbert method have been largely replaced by better methods.
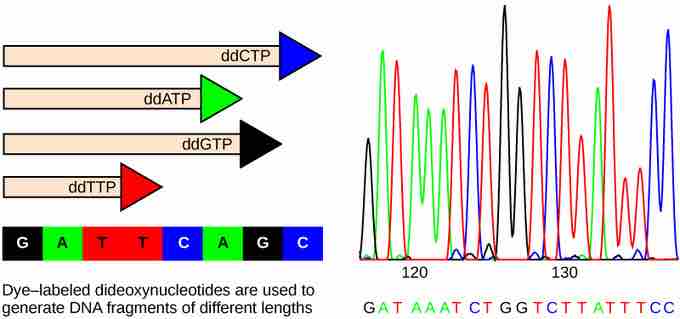
Sanger Method
In Frederick Sanger's dideoxy chain termination method, fluorescent-labeled dideoxynucleotides are used to generate DNA fragments that terminate at each nucleotide along the template strand. The DNA is separated by capillary electrophoresis on the basis of size. From the order of fragments formed, the DNA sequence can be read. The smallest fragments were terminated earliest, and they come out of the column first, so the order in which different fluorescent tags exit the column is also the sequence of the strand. The DNA sequence readout is shown on an electropherogram that is generated by a laser scanner.
Sanger Sequencing
The Sanger method is also known as the dideoxy chain termination method. This sequencing method is based on the use of chain terminators, the dideoxynucleotides (ddNTPs). The dideoxynucleotides, or ddNTPSs, differ from deoxynucleotides by the lack of a free 3' OH group on the five-carbon sugar. If a ddNTP is added to a growing DNA strand, the chain is not extended any further because the free 3' OH group needed to add another nucleotide is not available. By using a predetermined ratio of deoxyribonucleotides to dideoxynucleotides, it is possible to generate DNA fragments of different sizes when replicating DNA in vitro.
A Sanger sequencing reaction is just a modified in vitro DNA replication reaction. As such the following components are needed: template DNA (which will the be DNA whose sequence will be determined), DNA Polymerase to catalyze the replication reactions, a primer that basepairs prior to the portion of the DNA you want to sequence, dNTPs, and ddNTPs. The ddNTPs are what distinguish a Sanger sequencing reaction from just a replication reaction. Most of the time in a Sanger sequencing reaction, DNA Polymerase will add a proper dNTP to the growing strand it is synthesizing in vitro. But at random locations, it will instead add a ddNTP. When it does, that strand will be terminated at the ddNTP just added. If enough template DNAs are included in the reaction mix, each one will have the ddNTP inserted at a different random location, and there will be at least one DNA terminated at each different nucleotide along its length for as long as the in vitro reaction can take place (about 900 nucleotides under optimal conditions.)
The ddNTPs which terminate the strands have fluorescent labels covalently attached to them. Each of the four ddNTPs carries a different label, so each different ddNTP will fluoresce a different color.
After the reaction is over, the reaction is subject to capillary electrophoresis. All the newly synthesized fragments, each terminated at a different nucleotide and so each a different length, are separated by size. As each differently-sized fragment exits the capillary column, a laser excites the flourescent tag on its terminal nucleotide. From the color of the resulting flouresence, a computer can keep track of which nucleotide was present as the terminating nucleotide. The computer also keeps track of the order in which the terminating nucleotides appeared, which is the sequence of the DNA used in the original reaction.
Second Generation and Next-generation Sequencing
The Sanger and Gilbert methods of sequencing DNA are often called "first-generation" sequencing because they were the first to be developed. In the late 1990s, new methods, called second-generation sequencing methods, that were faster and cheaper, began to be developed. The most popular, widely-used second-generation sequencing method was one called Pyrosequencing.
Today a number of newer sequencing methods are available and others are in the process of being developed. These are often called next-generation sequencing methods. The most widely-used sequencing method currently is one called Illumina sequencing (after the name of the company which commercialized the technique), but numerous competing methods are in the developmental pipeline and may supplant Illumina sequencing.
In Illumina sequencing, up to 500,000,000 separate sequencing reactions are run simultaneously on a single slide (the size of a microscope slide) put into a single machine. Each reaction is analyzed separately and the sequences generated from all 500 million DNAs are stored in an attached computer. Each sequencing reaction is a modified replication reaction involving flourescently-tagged nucleotides, but no chain-terminating dideoxy nucleotides are needed.
When the human genome was first sequenced using Sanger sequencing, it took several years, hundreds of labs working together, and a cost of around $100 million to sequence it to almost completion. Next generation sequencing can sequence a comparably-sized genome in a matter of days, using a single machine, at a cost of under $10,000. Many researchers have set a goal of improving sequencing methods even more until a single human genome can be sequenced for under $1000.
Shotgun Sequencing
Sanger sequence can only produce several hundred nucleotides of sequence per reaction. Most next-generation sequencing techniques generate even smaller blocks of sequence. Genomes are made up of chromosomes which are tens to hundreds of millions of basepairs long. They can only be sequenced in tiny fragments and the tiny fragments have to put in the correct order to generate the uninterrupted genome sequence. Most genomic sequencing projects today make use of an approach called whole genome shotgun sequencing.
Whole genome shotgun sequencing involves isolating many copies of the chromosomal DNA of interest. The chromosomes are all fragmented into sizes small enough to be sequenced (a few hundred basepairs) at random locations. As a result, each copy of the same chromosome is fragmented at different locations and the fragments from the same part of the chromosome will overlap each other. Each fragment is sequenced and sophisticated computer algorithms compare all the different fragments to find which overlaps with which. By lining up the overlapped regions, a process called tiling, the computer can find the largest possible continuous sequences that can be generated from the fragments. Ultimately, the sequence of entire chromosomes are assembled.
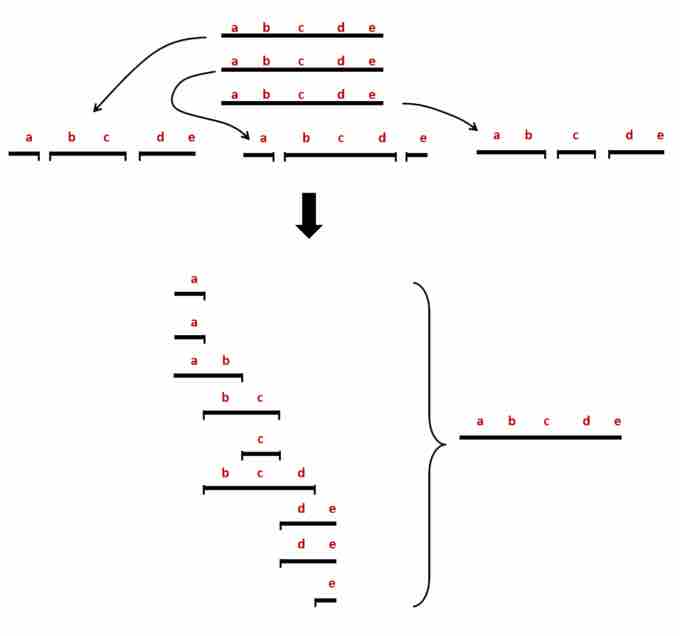
Whole genome shotgun sequencing.
In shotgun sequencing, multiple copies of the same chromosome are isolated and then fragmented in random locations. The different copies of the chromosome end up generating different length fragments. When the complete collection of fragments has been sequenced, comparing the sequences of all the fragments will reveal which fragments have ends that overlap with other fragments. The complete sequence from one end of the original DNA to the other can be assembled by following the sequence from the first overlapping fragment to the last.
Genome sequencing will greatly advance our understanding of genetic biology. It has vast potential for medical diagnosis and treatment.