In living organisms most of the biological functions are mediated by complex multi-component protein machineries and network activities. The protein complexes formed could be stable (proteins interact for a prolonged period of time) or transient (proteins interact for a brief period of time). Molecular studies are necessary to dissect the constituents of these protein complexes and identify the domains through which a protein interacts with another. Understanding how proteins are physically connected reveals clues about their structure and function and makes them an ideal target for drug therapy. Several methodologies exist to study the interaction of proteins in vivo. The most widely employed tools are the yeast two-hybrid system and affinity purification coupled to mass spectrometry. Datasets obtained from such tools are further analyzed using computational methods to draw a map of protein connectivity and achieve system level understanding of a microorganism. The complete map of protein interactions that can occur in a living organism is called the interactome.
The Yeast Two-hybrid System
The yeast two-hybrid screening system is an effective and quick tool for the in vivo study of protein–protein interaction both in prokaryotes and eukaryotes. The method consists of splitting a yeast transcription factor into its binding domain and activation domain, fusing the binding domain to one protein of interest (the bait) and the activation domain to another protein of interest (the prey), and reconstituting the activity of the transcription factor by bringing the two domains back into physical proximity. In the absence of an interaction the domains remain distant, preventing a detectable output. If the two proteins do interact the bait recruits the prey to a specific cellular location where it can stimulate a detectable output (e.g., gene activation). This experimental approach measures direct physical interaction between proteins and is called a binary method .
Affinity Purification Coupled to Mass Spectrometry
Affinity purification of protein complexes coupled to mass spectrometry is carried out as follows: a specific protein (the bait) is manipulated to express an affinity tag. The tag serves as a tool to purify the bait protein and associated proteins by affinity chromatography. Purified protein complexes are then resolved on native gels and discrete protein bands are excised and digested into small peptide fragments by trypsin.
Peptides are identified using mass spectrometry methods. The identity of the protein associated with a given bait protein is determined by comparing its peptide fingerprint against available databases. This method allows for the identification and quantification of direct binding partners and secondary interacting proteins, and assigns them into protein networks. This experimental approach measures physical interactions between groups of proteins without distinguishing whether they are direct or indirect and is termed co-complex method. Results collected from binary and co-complex experiments are documented into a database. There are many databases accessible online that allow for protein clustering by function and nature of interaction and provide a rich framework for biomedical research.
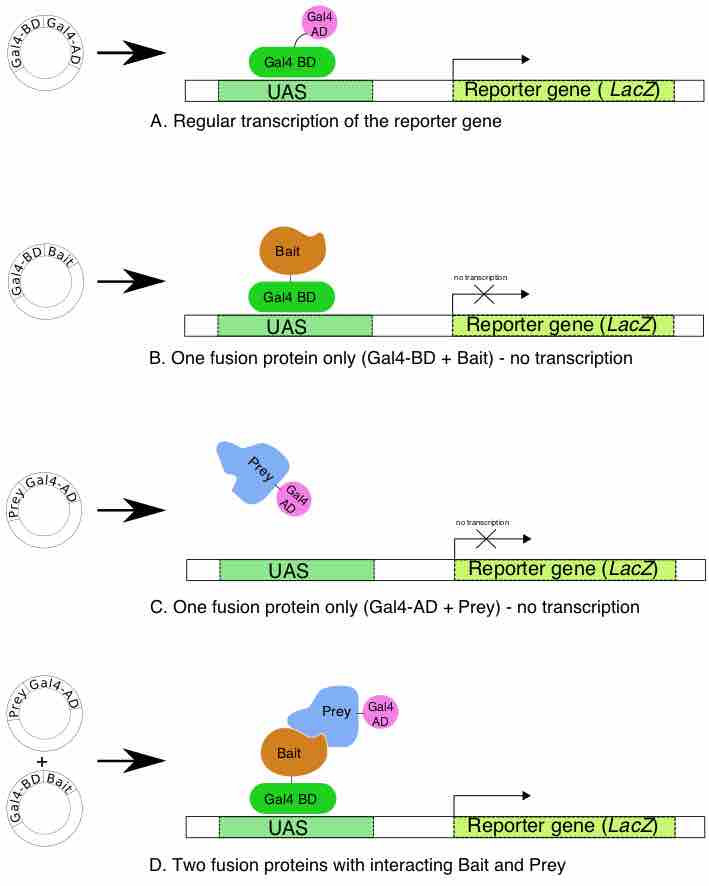
The Yeast two-hybrid method
Principle of the bait and prey method for the study of protein-protein interaction.