Polycystic Kidney Disease In Adults
- Article Author:
- Nancy Finnigan
- Article Editor:
- Stephen Leslie
- Updated:
- 8/10/2020 9:26:22 PM
- For CME on this topic:
- Polycystic Kidney Disease In Adults CME
- PubMed Link:
- Polycystic Kidney Disease In Adults
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a hereditary disorder of the kidneys characterized by markedly enlarged kidneys with extensive cyst formation throughout. [1] These cysts progressively enlarge with age, as kidney function gradually declines. The diagnosis of ADPKD is based on family history and ultrasonographic evaluation. In as many as 25% of patients with ADPKD, no family history is identified, which may be related to subclinical disease or a new genetic mutation in about 5% of cases. Patients with ADPKD typically progress to end-stage renal disease (ESRD) by the fifth or sixth decade of life. The rate of progression of ADPKD is related directly to kidney volume. Therapies aim to slow the decline in renal volume to delay progression.[2][3][4]
Screening family members for ADPKD has been controversial in the past. Prior to 2017, due to the lack of therapy to prevent progression to ESRD, screening of asymptomatic family members has not been routinely performed. With the addition of tolvaptan as a treatment option, early screening of young adults at risk for the disease would provide the opportunity for early treatment.
Etiology
ADPKD is due to an abnormality on chromosome 16 (PKD1 locus) or chromosome 4 (PKD2 locus). PKD1 mutations comprise about 78% of ADPKD cases, while PKD2 mutations comprise about 14% of cases. The remaining cases have no identifiable mutations. PKD1 patients tend to progress to ESRD at an earlier age than PKD2 patients (mean age 54.3 versus 74.0).
Epidemiology
ADPKD occurs in approximately 1 in every 1000 births, with a global prevalence of 10 per 10,000. As many cases have a benign prognosis, it is likely that less than half of these patients are ever diagnosed.
Pathophysiology
The PKD1 and PKD2 genes encode the proteins polycystin-1 and polycystin-2, respectively. These polycystins are integral membrane proteins and are found in renal tubular epithelia. It is postulated that abnormalities in polycystin-1 impair cell-cell and cell-matrix interactions in the renal tubular epithelia, while abnormalities in polycystin-2 impair calcium signaling in the cells.[5][6][7]
The resultant changes in renal pathophysiology include hematuria (often gross), a concentrating defect (resulting in polyuria and increased thirst), mild proteinuria, nephrolithiasis (in about 25% of ADPKD patients), flank pain, and abdominal pain. Cyst rupture, hemorrhage, and infection are common complications. Progressive renal decline often results in end-stage renal disease.
Histopathology
The diagnosis of ADPKD is based on ultrasonographic criteria in patients with a positive family history. Routine biopsies for microscopic examinations are not performed.
History and Physical
Patients with ADPKD may present with a variety of medical conditions, including hypertension, flank pain, abdominal masses, urinary tract infections, renal failure, nephrolithiasis, and cerebrovascular accidents. Hypertension is the most prevalent initial clinical presentation, occurring in 50% to 70% of cases, and is the most common feature directly associated with the rate of decline to ESRD and cardiovascular complications.
Multiple extra-renal manifestations are often present. Cerebral aneurysms occur in about 5% of young adults, and as many as 20% of patients over the age of 60. The risk of a cerebral aneurysm or subarachnoid hemorrhage is highest in patients with a family history of the same.
Extrarenal cysts are common in ADPKD. Hepatic cysts are often noted in these patients, and the prevalence increases with age. As many as 94% of patients over the age of 35 have been reported to have hepatic cysts. Total cyst prevalence and volume is higher in women versus men. Hepatic cysts in ADPKD patients rarely cause liver dysfunction. Rarely, patients develop pain from an acute cyst infection or hemorrhage. In addition, about 7% to 36% of ADPKD patients develop pancreatic cysts, with a higher prevalence in ADPKD patients with PKD2 mutations.
Cardiac valvular disease has been noted in 25% to 30% of ADPKD patients. Cardiovascular complications, particularly cardiac hypertrophy and coronary artery disease, are the leading causes of death in patients with ADPKD.
Additional complications include colonic diverticula, which are noted in ADPKD patients requiring maintenance dialysis. Abdominal wall hernias are noted in as many as 45% of ADPKD patients, likely related to additional abdominal girth related to increased kidney size.
Evaluation
The diagnosis of ADPKD is made based on ultrasonographic criteria. The criteria for ultrasonographic diagnosis of individuals with a positive family history with unknown genotype (as is usually the case) is as follows:
- In patients ages 15 to 39 years, at least three unilateral or bilateral renal cysts
- In patients ages 40 to 59 years, at least two cysts in each kidney
- In patients age 60 years or older, at least four cysts in each kidney
While ultrasound is the only study required to make a definitive diagnosis, patients are routinely evaluated with a basic metabolic profile and urinalysis to determine the extent of any renal insufficiency.
Treatment / Management
Early management of hypertension is pivotal in reducing cardiovascular mortality, the incidence of left ventricular hypertrophy, mitral regurgitation, and to slow the progression of renal failure. While the target blood pressure in ADPKD patients has yet to be established, the HALT-PKD Study A noted that in early stages of ADPKD with preserved renal function, blood pressure management with systolic blood pressure less than 110 mmHg was strongly associated with significant reductions in the rate of total kidney volume growth, albuminuria, and left ventricular mass index. ACE inhibitors and ARBs are the mainstays of therapy, with beta-blockade and calcium-channel blockers as second-line therapy. As third-line therapy, thiazides are preferred in patients with normal renal function, while loop diuretics are preferred in patients with impaired renal function.[8][9][10][11]
Screening for a cerebral aneurysm is recommended at the time of ADPKD diagnosis in patients that are high risk (those with a family history of an aneurysm or intracranial hemorrhage in a first-degree relative).
Smoking cessation and hypertension management are most prudent, as smoking and high blood pressure increase the risk of a cerebral aneurysm. Of note, RAS blockade and statin use decrease the cerebral aneurysm rate.
Tolvaptan has demonstrated a slower decline than placebo in the eGFR over a one year period in patients with late-stage chronic kidney disease but is associated with elevations of bilirubin and alanine aminotransferase levels.
Multiple additional therapies have been studied in the effort to prevent progression of ADPKD. Dietary sodium restriction, which was part of the HALT-PKD trial, was shown to possibly reduce renal progression, as sodium excretion was associated with an increased risk of kidney growth and eGFR decline.
Statins have shown some benefit in all-cause chronic kidney disease and have suggested benefit in ADPKD patients. Statins are deployed in chronic kidney disease patients as progressive renal failure is a coronary heart disease equivalent.
Mammalian target of rapamycin (mTOR) inhibitors such as sirolimus have been studied but have not shown any benefit on renal outcomes. Diuretics, such as amiloride, have also been tried to decrease cyst volume without measurable improvement in renal function. Protein restriction has provided variable results.
Octreotide has also been studied in ADPKD and has shown a non-significant slowing of renal function decline with possible attenuation of the effect after 2 years.[12][13]
Differential Diagnosis
The differential diagnosis of renal cysts in adults includes several conditions, including simple renal cysts, complex renal cysts, localized cystic disease, malignancy, and acquired cystic disease of the kidney. It is important to distinguish disorders that may have a malignant potential, such as complex cysts, acquired the cystic disease, and underlying malignancy. A defining feature of ADPKD is marked bilateral, renal enlargement.
Prognosis
ADPKD patients reach ESRD, on average, in the fifth or sixth decade of life. ADPKD accounts for about 10% of all ESRD cases. Patients with hypertension and larger kidney size tend to have a worse prognosis. A family history of the disease may be predictive of the specific mutation and predictive of the patient's course.
Pearls and Other Issues
Autosomal dominant polycystic kidney disease often presents in young adults and is responsible for about 10% of all ESRD cases. Early intervention with optimal blood pressure management along with emerging medical therapies such as tolvaptan are the mainstays of treatment.
Enhancing Healthcare Team Outcomes
The management of patients with ADPKD is optimal with an interprofessional team. The key to management is the control of blood pressure and slow down the progression towards end stage renal disease. Healthcare workers who follow these patients should closely monitor the blood pressure and renal function at regular intervals.
ACE inhibitors and ARBs are the mainstays of therapy, with beta-blockade and calcium-channel blockers as second-line therapy. As third-line therapy, thiazides are preferred in patients with normal renal function, while loop diuretics are preferred in patients with impaired renal function.
These patients also need to be screened for a cerebral aneurysm. Patients should be urged to quit smoking and remain compliant with their antihypertensive medications.
Multiple additional therapies have been studied in the effort to prevent the progression of ADPKD. Dietary sodium restriction, which was part of the HALT-PKD trial, was shown to possibly reduce renal progression, as sodium excretion was associated with an increased risk of kidney growth and eGFR decline.
Statins have shown some benefit in all-cause chronic kidney disease and have suggested benefit in ADPKD patients. Statins are deployed in chronic kidney disease patients as progressive renal failure is a coronary heart disease equivalent.
For patients who fail to control their blood pressure, the prognosis is poor. (Level V)
(Click Image to Enlarge)

CT Abdomen Autosomal Dominant Polycystic Kidney Disease
Contributed by Scott Dulebohn, MD
(Click Image to Enlarge)

Polycystic Kidney
Contributed by Dr. Michael Lambert
(Click Image to Enlarge)
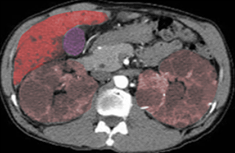
AutosomalPolycystickidney
Image courtesy S Bhimji MD