Anatomy, Abdomen and Pelvis, Kidney Collecting Ducts
- Article Author:
- Robert McMahon
- Article Author:
- Dana Penfold
- Article Editor:
- Khalid Bashir
- Updated:
- 7/27/2020 9:18:07 PM
- For CME on this topic:
- Anatomy, Abdomen and Pelvis, Kidney Collecting Ducts CME
- PubMed Link:
- Anatomy, Abdomen and Pelvis, Kidney Collecting Ducts
Introduction
Renal collecting ducts are microscopic passages that connect to multiple nephrons. Tubular fluid passes through the collecting ducts to reach the calyces and renal pelvis. While traveling through the collecting ducts, the composition of tubular fluid undergoes alterations. These alterations pertain primarily to reabsorption and secretion of electrolytes, acid, and water. Different cell types line the collecting duct lumen to mediate the alterations. These cell types are under the regulation of chemical messengers originating from both different organ systems and the kidneys themselves.
Structure and Function
The collecting ducts can subdivide into three anatomical segments: cortical, outer medullary, and inner medullary. At each of these segments, collecting ducts connect with distal aspects of nephrons called collecting tubules. A lobule is comprised of a collecting duct and the group of nephrons that it drains.[1] Medullary rays extend into the cortex from the medulla and are composed of collecting ducts and straight tubules.[1] Collecting ducts drain into the papillary ducts and subsequent minor calyces via papillae at the apices of the pyramids, the area cribrosa.[1] Once the tubular fluid reaches a collecting duct, three cell types predominate. They are the principal cells, type A intercalated cells, and type B intercalated cells. Each cell type has characteristically distinct apical and basolateral plasma membrane proteins.[2]
Principal cells contain sodium (ENaC) and potassium channels on their apical surfaces. Aldosterone promotes the expression of each channel, increasing both sodium reabsorption and potassium secretion. Atrial natriuretic peptide (ANP) opposes these aldosterone-mediated effects. Also, principal cells may express apical aquaporin two channels in response to stimulation by antidiuretic hormone (ADH). Increased expression of aquaporin 2 promotes water reabsorption, which concentrates the tubular fluid. The basolateral surfaces of principal cells contain Na-K-ATPases that create gradients for promoting sodium reabsorption and potassium secretion.[3]
Type A intercalated cells primarily function to secrete acid into the tubular fluid. This function takes place with apical H-ATPases and H/K-ATPases. Intracellular carbonic acid dissociates to form a hydrogen ion (conjugate acid) and bicarbonate (conjugate base). Bicarbonate enters the circulation via basolateral anion exchanger 1, while hydrogen ions get secreted through the aforementioned apical ATPases. Secreted hydrogen ions bind to ammonia and hydrogen phosphate to become substances that are generally not reabsorbed. Aldosterone can potentiate this process by increasing the expression of H-ATPase.[4]
Type B intercalated cells can be thought of as the base-secreting equivalents of type A intercalated cells. Like in type A intercalated cells, carbonic acid dissociates into a hydrogen ion and bicarbonate. Bicarbonate is secreted utilizing an apical anion exchanger called pendrin. Hydrogen ions enter the circulation via basolateral H-ATPase pumps.[4]
Cells of the inner medullary cortical duct segment can express urea channels. This capacity contributes to the overall concentrating effect ADH has on tubular fluid. In the presence of ADH, expression of apical urea transporter-A1 and basolateral urea transporter-A3 increases, which allows urea to enter the inner medullary interstitial space, increasing its concentration. This effect enhances water reabsorption by potentiating passive sodium and chloride reabsorption in the thin ascending limb of the loop of Henle.[5]
Embryology
Nephrogenesis develops in three consecutive phases: the pronephros, mesonephros, and metanephros. By the end of the fourth week of gestation, the mesonephros derives from the intermediate mesoderm and is composed of excretory tubules received by collecting ducts, termed mesonephric or Wolffian ducts.[6][1] Collecting ducts arise from the ureteric bud, which is also the progenitor for the calyces, renal pelvis, ureters, and bladder trigone. The ureteric bud invades the metanephric mesenchyme and undergoes a series of tightly regulated branching events. Branch tips will eventually interact with cap mesenchyme cells to promote nephron development and attachment to the collecting system. GDNF/RET, FGF, Six1/Sall1, and Wnt signaling pathways are involved in the various stages of ureteric bud growth, invasion, and branching.[7] The mesonephric duct is stimulated by the interaction between its tyrosine kinase inhibitor RET and glial-derived neurotrophic factor (GDNF) and between its tyrosine kinase inhibitor MET and hepatocyte growth factor (HGF) to evaginate and form the ureteric bud. Wilms tumor 1 (WT1) is a transcription factor expressed by the metanephric mesenchyme and regulates the production of GDNF and HGF.[1][6][8]
Blood Supply and Lymphatics
Both of the renal arteries branch off of the aorta and divide into five segmental arteries: the posterior segmental artery, superior segmental artery, anterosuperior segmental artery, anteroinferior segmental artery, and inferior segmental artery. These segmental arteries branch into interlobar arteries which continue to become the arcuate arteries. As collecting ducts span both the cortex and medulla, more than one aspect of the renal vasculature may supply them. In the cortex, interlobular arteries branch perpendicularly off of arcuate arteries, which lie at the borders of cortical and medullary tissue. Arcuate arteries give rise to radial arteries. In the medulla, vasa recta capillaries are present (specifically around loops of Henle). The renal veins and branches follow the path of the renal arteries.[9][10][11]
Lymph from the kidneys primarily passes through the hilar lymphatic route. This route involves lymphatic vessels traveling along the renal vasculature to reach aortic and caval nodes eventually. From these nodes, lymph proceeds to the thoracic duct.[12]
Nerves
The kidneys are innervated by autonomic fibers that influence vascular, tubular, and excretory functions. Postganglionic nerves from the renal plexus travel along renal vasculature to enter the parenchyma. Sympathetic innervation comes from spinal cord levels T11-L3 and innervates the nephrons and renal vasculature completely. It is concentrated most heavily around the afferent arterioles, thick ascending limbs, and distal convoluted tubules. The vagus nerve contributes parasympathetic fibers.[13][1]
Muscles
Clinically, the musculature and kidneys are connected primarily through creatinine. Creatinine is a muscle breakdown product that has been used clinically to evaluate kidney function for decades. In damaged muscles (e.g., rhabdomyolysis), myoglobin can enter the bloodstream and exert nephrotoxic effects. On the other hand, healthy muscles are believed to protect against inflammation-mediated kidney deterioration through several mechanisms. Macroscopically, the kidneys are anterior to the psoas muscles within Gerota’s fascia.[14][15][16][1]
Physiologic Variants
Liddle syndrome: Liddle syndrome is a rare autosomal dominant disorder caused by gain-of-function mutations in genes encoding ENaC subunits. Symptoms include a triad of early-onset hypertension, hypokalemia, and metabolic alkalosis. Normal to low aldosterone activity and lack of response to spironolactone distinguish this syndrome from hyperaldosteronism. The clinician can achieve a definitive diagnosis through genetic testing. Treatment primarily involves maintaining a low sodium diet and the use of potassium-sparing diuretics called ENaC blockers (elaborated on in the Clinical Significance section).[17]
Carbonic anhydrase II deficiency: In the collecting ducts, carbonic anhydrase II (CAII) mediates the formation of carbonic acid from water and carbon dioxide. Carbonic acid then dissociates into a hydrogen ion and bicarbonate, which are handled differently by type A and type B intercalated cells (as described in the Structure and Function section). CAII deficiency is an autosomal recessive disorder that manifests clinically as osteopetrosis, cerebral calcification, and renal tubular acidosis (RTA). The renal tubular acidosis that manifests may be proximal (type II), distal (type I), or combined (type III). No specific treatment (aside from symptomatic) for CAII deficiency currently exists.[18][19]
Surgical Considerations
Collecting duct carcinoma (CDC) is an aggressive form of renal cell carcinoma. CDC represents fewer than 2% of renal masses and usually carries a poor prognosis. Alterations in various genes, including NF2, SETD2, SMARCB1, and CDKN2A, have been identified in CDC cases. Patients with CDC generally undergo cytoreductive nephrectomies, which have been shown to improve survival outcomes (particularly when combined with chemotherapy/radiation).[20][21][22]
Clinical Significance
In addition to the conditions already mentioned, many clinical considerations involve the collecting ducts:
Nephrogenic diabetes insipidus (NDI): NDI is the inability of collecting ducts to concentrate tubular fluid due to an intrinsic renal complication. Such a complication can be acquired or genetic. Acquired causes include drug toxicities, electrolyte imbalances, and systemic disorders (e.g., autoimmune, amyloidosis, sarcoidosis, etc.). The most common cause of drug-associated NDI is due to lithium use. Lithium is believed to downregulate aquaporin 2 expression in principal cells. Genetic causes involve loss-of-function mutations in genes encoding either vasopressin receptor 2 (the receptor for ADH in the collecting ducts) or aquaporin 2. Clinically, NDI manifests as polyuria due to the excretion of excess water. This loss of fluid leads to secondary polydipsia. Diagnosis can be confirmed with a water deprivation test and a lack of response to desmopressin. Improvement with desmopressin would indicate central diabetes insipidus, which is due to impaired ADH release from the posterior pituitary gland. Treatment options for NDI include addressing reversible secondary causes, thiazide diuretics (with the optional addition of potassium-sparing diuretics), and low solute diets.[23]
Syndrome of inappropriate antidiuretic hormone secretion (SIADH): SIADH is characterized by excessive fluid retention due to aberrantly high ADH activity. SIADH can have many etiologies, including neoplasia, certain drugs, intracranial disorders, and pulmonary disorders. Clinically, SIADH appears as euvolemic hyponatremia with initially increased sodium excretion in the urine. Increased retention of solute-free water from the collecting duct increases circulating volume, diluting the serum sodium. However, this increase in circulating volume prompts a decrease in the renin-angiotensin-aldosterone system (RAAS) activity, which restricts sodium and water reabsorption by principal cells. Thus, the overall effect is the maintenance of normal circulating volume. Sodium excretion will eventually match sodium intake again with the establishment of a new steady state. Therapeutic strategies mainly involve addressing underlying etiologies and preventing severe hyponatremia. Care should be taken not to correct hyponatremia too rapidly, as this can lead to osmotic demyelination syndrome.[24][25][26]
Potassium-sparing diuretics: Potassium-sparing diuretics interfere with sodium reabsorption at principal cells. If less sodium reabsorption occurs, the lumen-negative transepithelial potential difference driving subsequent potassium secretion gets reduced. The result is increased excretion of sodium and water and decreased excretion of potassium. There are two groups of potassium-sparing diuretics: mineralocorticoid receptor (MR) antagonists and ENaC blockers.[27]
- MR antagonists: The intracellular MR is where aldosterone binds to enhance sodium reabsorption and potassium secretion in principal cells. Examples of MR antagonists are spironolactone and eplerenone. Spironolactone's primarily use is for hypertension, but it can be a adjunct to conventional heart failure regimens. To lessen the risk of hyperkalemia, a thiazide diuretic can be included to lower the spironolactone dosage. Despite the clinical efficacy of spironolactone, it can exert potentially distressing antiandrogenic effects in men like gynecomastia and erectile dysfunction. Eplerenone is understood to have fewer of these antiandrogenic side effects while maintaining clinical efficacy. [28]
- ENaC blockers: ENaC blockers are generally less efficacious for treating hypertension than MR antagonists. As such, the administration of ENaC blockers like amiloride and triamterene is usually with thiazide diuretics. As with MR antagonists, hyperkalemia is a potential side effect. Amiloride can also cause mild gastrointestinal discomfort. Triamterene use can potentially predispose hyperuricemia and kidney stone formation.[28]
Renal tubular acidosis: Types I, III, and IV RTA involve the collecting ducts. Type I RTA is classically an inability of type A intercalated cells to secrete hydrogen ions and reabsorb potassium ions, resulting in hyperchloremic (or normal anion gap) metabolic acidosis and hypokalemia with urine pH greater than 5.5. Type I RTA can be caused by genetic mutations, systemic (e.g., autoimmune, amyloidosis, sarcoidosis, etc.) diseases, and nephrotoxic drugs. Compounds like sodium bicarbonate and potassium citrate can correct the acid and potassium imbalances, respectively, but underlying causes should be addressed if present. Alternatively, type IV RTA is a consequence of aldosterone deficiency or resistance; this manifests primarily as hyperchloremic metabolic acidosis with hyperkalemia. Treatment strategies for type IV RTA include restricting potassium intake and administering bicarbonate if the acidosis necessitates it. Type III RTA is a very rare condition that manifests with characteristics of both type I and type II (not discussed in this review) RTA. Type III RTA can occur as a consequence of acetazolamide misuse or CAII deficiency.[29][30][31]
Hyperaldosteronism: Aberrantly increased aldosterone activity can result from primary or secondary causes. Primary causes include bilateral adrenal hyperplasia and aldosterone-producing adrenal tumors, which produce aldosterone regardless of RAAS activity. Classically, primary hyperaldosteronism manifests as hypervolemia, hypertension, hypokalemia, and metabolic alkalosis. Hypervolemia and hypertension are due to excessive sodium and water retention by principal cells. The alkalosis is due to excessive acid secretion by type A intercalated cells. While hypokalemia is considered a classic symptom, it is estimated to appear in fewer than 40% of cases. Secondary hyperaldosteronism is due to overactivation of RAAS. Many etiologies can stimulate RAAS activity, making a definitive symptom profile for secondary hyperaldosteronism difficult to establish. Treatment options include adrenalectomy, MR antagonists, and addressing any secondary causes.[32][33][34]
Cushing’s syndrome: Cortisol can exert mineralocorticoid effects at sufficiently high levels. In cells containing MR, an enzyme called 11-beta-HSD2 (11B-HSD2) metabolizes cortisol before it can bind to MR. Excess cortisol can overcome cellular 11B-HSD2 capacity, allowing cortisol to bind to MR. Binding to MR can lead to excessive sodium and water retention in the collecting ducts. This effect contributes to the hypertension seen in Cushing syndrome. Treatment involves addressing the cause of the excess cortisol (i.e., tapering corticosteroid use, resection of an adrenal tumor, etc.).[35]
Other Issues
In the last few years, many investigative avenues involving the collecting ducts have been subject to research. One study from 2017 characterized the role of TFCP2L1, a transcription factor, in mediating collecting duct development. Another study from 2018 demonstrated the development of tubulointerstitial fibrosis with the suppression of microRNAs within collecting duct cells. Most recently, a 2019 study found that tamoxifen downregulated collecting duct aquaporin 2 in an ADH-independent manner. These recent scholarly endeavors demonstrate the scientific and clinical necessities of understanding the renal collecting ducts.[36][37][38]
(Click Image to Enlarge)
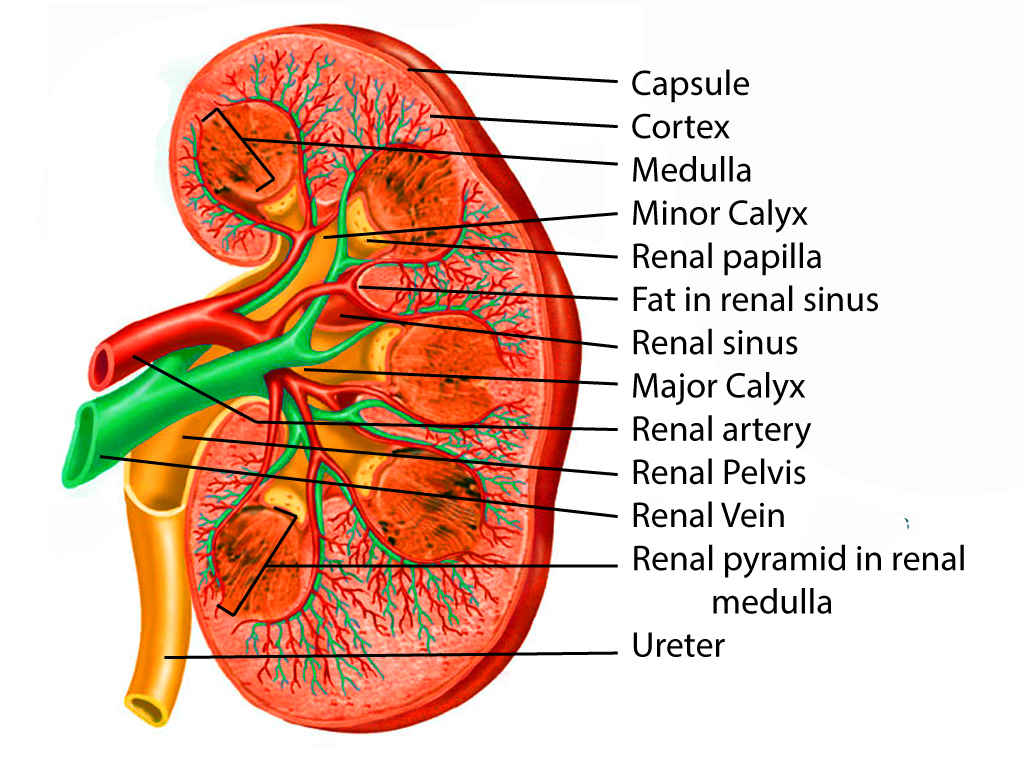
Kidney anatomy
Contributed by Scott Dulebohn, MD
(Click Image to Enlarge)
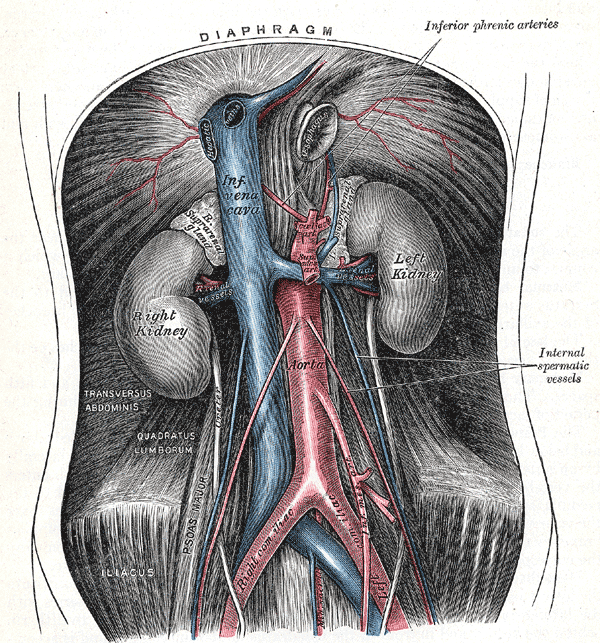
The abdominal aorta and its branches. Diaphragm, Inferior Phrenic arteries, Internal spermatic vessels, Left and Right Kidney, Inferior vena cava, Aorta, Transversus Abdominis, Quadratus lumborum, Right and left Iliac
Contributed by Gray's Anatomy Plates
(Click Image to Enlarge)

Normal urine production
Image courtesy S Bhimji MD
(Click Image to Enlarge)

Intercalated cell
Image courtesy: https://commons.wikimedia.org/wiki/File:Alpha_Intercalated_Cell_Cartoon.svg