Maple Syrup Urine Disease
- Article Author:
- Syed Adeel Hassan
- Article Editor:
- Vikas Gupta
- Updated:
- 10/5/2020 10:03:41 AM
- For CME on this topic:
- Maple Syrup Urine Disease CME
- PubMed Link:
- Maple Syrup Urine Disease
Introduction
Maple syrup urine disease (MSUD) was first described as a rapid onset of Menkes' neurodegenerative disease in 1954.[1] It is a defect of metabolism due to abnormal activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. This complex is responsible for the breakdown of branched-chain amino acids:
- Leucine
- Isoleucine
- Valine
The underlying defect in the BCKAD complex disrupts the metabolism of branched-chain amino acids, which leads to an accumulation of branched-chain amino acids (BCAAs) in the plasma and their respective branched-chain ketoacids in the urine.[2] It classically manifests in the neonatal period with failure to thrive, delayed developmental milestones, feeding difficulties, and a maple syrup odor in the urine or cerumen.[2] Treatment consists of close metabolic monitoring and dietary restriction of branched-chain amino acids. If left untreated, irreversible neurological damage and metabolic catastrophe ensue. Good clinical outcomes can be expected if management is initiated early.
Etiology
Four subunits comprise the BCKAD multienzyme complex. These subunits include dihydrolipoamide dehydrogenase (E3 subunit) and BCKAD decarboxylase (E1 subunit) bound to dihydrolipoyl acyltransferase (E2 subunit).[1] Two regulatory subunits (BCKAD kinase and BCKAD phosphatase) are also associated with the E2 subunit's core.[1] Furthermore, the E1 subunit is composed of E1alpha and two E1beta subunits. The metabolic disorder occurs due to a decreased activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex which is located within the mitochondria. The defect is transmitted in an autosomal recessive pattern. There are five main clinical phenotypes of maple syrup urine disease.[3]
Epidemiology
Maple syrup urine disease involves males and females equally. It has an estimated worldwide incidence of 1 case per 185,000 live births.[1] Higher occurrences have been noted in populations with a higher rate of consanguinity. In the Ashkenazi Jewish population, the incidence is estimated at 1 in 26,000 live births. In Mennonites, maple syrup urine disease occurs with an incidence of 1 in 380 newborns. This is often termed as a founder's effect in the BCKDHA (E1a) gene (c.1312T>A). In Portuguese gypsies, 1.4% of cases occur due to the homozygous deletion (117delC) of the BCKDHA gene. It can be estimated as 1 case per 71 births.[4]
Pathophysiology
Branched-chain ketoacid dehydrogenase (BCKAD) is located within the inner mitochondrial membrane of various tissues such as skeletal muscle, liver, kidney, and the brain. It is composed of three catalytic subunits (E1, E2, and E3). Together with branched-chain amino acid transaminase, it helps mediate catabolism of branched-chain amino acids (BCAA). In the presence of thiamin pyrophosphate, E1 decarboxylates the alpha ketoacids. The lipoic acid residue in E2 transfers the acyl group from E1 to CoA. The E3 subunit helps reoxidize the lipoic acid residue in E2. The activity of branched-chain ketoacid dehydrogenase is further regulated by BCKAD phosphatase and BCKAD kinase. Therefore, within the mitochondria, branched-chain amino acids are first converted into their respective alpha-ketoacids by the enzyme branched-chain amino acid transaminase. Their respective yielded alpha-ketoacids include alpha-ketoisocaproic acid, alpha-keto-beta-methyl valeric acid, and alpha-ketoisovaleric acid. Alpha-ketoacids are then oxidatively decarboxylated by the branched-chain ketoacid dehydrogenase complex. Consequently, alpha-ketoacids are further metabolized into intermediates such as isovaleryl-coenzyme A, alpha-methylbutyryl-CoA, and isobutyrl-CoA. These intermediates are then converted into succinyl-CoA, acetoacetate, and acetyl-CoA.[2] The branched-chain amino acids are essential amino acids with hydrophobic side chains and are found in protein-rich food. The catabolism of these amino acids is necessary to maintain various physiologic functions such as :
- Protein synthesis
- Gluconeogenesis
- Fatty acid synthesis
- Cholesterol synthesis
- Cellular signaling
In the brain, BCKAD helps metabolize BCAA to facilitate cerebral GABA and glutamate synthesis. The liver and kidney are responsible for the catabolism of 10% to 15% of BCAA.[5] Most BCAA transamination and oxidation occur in the skeletal muscle.[6] Maple syrup urine disease occurs due to a pathogenic defect in any BCKAD subunit resulting in elevated branched-chain amino acids and their corresponding alpha keto-acids. Accumulated BCAA and alpha-ketoacids manifests as a constellation of clinical symptoms due to dysfunction of the central nervous system, immune system, and skeletal muscle. Elevated leucine and alpha-ketoisocaproic acid levels notoriously cause neurochemical disturbances resulting in clinically apparent neurotoxicity. The transport of large neutral amino acids across the blood-brain barrier is drastically reduced due to interference from elevated leucine levels. As a result, the supply of tyrosine, phenylalanine, methionine, tryptophan, histidine, and valine to the brain.[3] As a consequence, brain growth and myelin synthesis are negatively impacted. The restricted supply of the amino acids leads to decreased neurotransmitters, such as dopamine, serotonin, norepinephrine, epinephrine, GABA, and glutamate. Alpha-ketoisocaproic acid levels of greater than 60 micromol/L negatively regulate transamination reactions within astrocytes.[3] This reversal accounts for low cerebral glutamate levels, which results in cognitive dysfunctions such as learning disabilities and memory loss. Furthermore, elevated leucine concentrations impair cell volume regulation. This results in decreased blood osmolarity, sodium concentrations, and increased intracellular water leading to cerebral edema.[7] In infants and children, decreased blood osmolarity can further precipitate brain herniation. Clinical evidence suggests that the neurotoxin alpha-ketoisocaproic acid contributes to the encephalopathic syndrome.[2] In patients with classic MSUD, decreased levels of glutamate and increased cerebral lactate levels indicate inhibition of the respiratory chain by alpha-ketoisocaproic acid.[3] The metabolic decompensations in MSUD lead to the activation of matrix metalloproteinases, resulting in the blood-brain barrier's breakdown.[8] Isoleucine metabolites are responsible for the maple syrup odor of the urine.
History and Physical
There are five clinical subtypes of MSUD. These include:
- The classic
- Intermediate
- Intermittent
- Thiamine responsive
- E3 deficient subtypes
They are clinically distinguished based on their clinical presentation, age of onset, and residual BCKAD enzyme activity.[1] There is no good correlation between genotype and phenotype. The clinical presentation relies on the BCKAD residual function.[2] However, phenotypic classification depends on the response to metabolic decompensation and leucine tolerance. The specific characteristics and the differences between the different types are shown in table 1.[2][3]
Evaluation
Evaluation of maple syrup urine disease requires prompt identification of clinical characteristics by a physician. In addition to the clinical features, biochemical testing and molecular testing play a significant role in disease phenotype evaluation.
Clinical Characteristics
Clinical characteristics depend on the underlying metabolic phenotype of MSUD. It is important to note the following manifestations:
- Maple syrup odor in cerumen is the first detectable clinical sign within the first 12 hours after birth.
- 2 to 3 days of age: Irritability, ketonuria, and poor feeding
- A worsening course of encephalopathy presenting as apnea, opisthotonus, and lethargy. Furthermore, fencing and bicycling movements can also develop by the age of 4 to 5 days.
- Coma and respiratory failure by the age of 7-10 days.
- Milder forms of the disease can present later in infancy/childhood as anorexia, stunted growth, and delayed milestones. Such cases will only develop encephalopathy and ketonuria during metabolic decompensation during an acute illness.
Diagnosis
Children and newborns should be tested for MSUD if the following are present:
- Sporadic encephalopathy and ketoacidosis
- Intermittent encephalopathy and ketoacidosis
- Encephalopathy and ketoacidosis during an underlying illness
- Encephalopathy and ketoacidosis following trauma or fasting
- Encephalopathy and ketoacidosis with a negative newborn screening result
Prenatal Diagnosis
Diagnosis requires the measurement of BCKAD enzyme activity in cultured chorion villus cells or amniocytes using mutational analysis. Branched-chain amino acid concentrations can also be measured in amniotic fluid. Pre-implantation diagnosis requires the identification of familial pathogenic variants. The preferred diagnostic method is molecular analysis.
Newborn Screening and Biochemical Evaluation
Since 1964, routine newborn screening has been performed for MSUD. The screening is performed in all 50 United States and is best performed within 24-48 hours after birth. It is performed with tandem mass spectrometry amino acid profiling (MS/MS). The process examines the fisher ratio and concentrations of leucine-isoleucine as a standard measure.[2] Additional laboratory studies need to be performed if elevated branched-chain amino acids are detected.[2] Tandem mass spectrometry readily detects classic MSUD. However, it may not detect milder forms of MSUD due to normal leucine levels. MS/MS does not possess the capability to distinguish amino acids with the same mass, such as hydroxyproline, leucine, isoleucine, and allo-isoleucine.[2] In such cases, a second-tier test such as liquid chromatography must be conducted to analyze allo-isoleucine on dry blood spots. Second-tier testing can help distinguish MSUD from hydroxyprolinemia. Additional laboratory studies include dinitrophenylhydrazine test, gas chromatography, liquid chromatography, BCKAD enzyme activity, and molecular testing.[2]
Plasma amino acid testing is the most important diagnostic test for MSUD. It is used to assess elevated levels of BCAAs and allo-isoleucine. Elevated allo-isoleucine levels (> 5 micromols/L) confer high specificity and sensitivity for diagnosis. Even with elevated leucine levels, allo-isoleucine levels are not detectable until the age of six days. The urinary organic acids can be detected using gas chromatography-mass spectrometry, dinitrophenylhydrazine tests, and urine test strips. Gas chromatography-mass spectrometry detects branched-chain ketoacids, which further support the diagnosis of MSUD. Branched-chain ketoacids usually follow after BCAA elevation and are detected 48-72 hours after birth.[2] The dinitrophenylhydrazine (DNPH) test can also be used to detect branched-chain ketoacids in the urine. DNPH reagent and urine are mixed in equal volumes. The sample is then observed for 10 minutes for any precipitation and color changes. Clear urine with no precipitate indicates a score of zero. Whereas, a yellow-white precipitate with opaque urine indicates a score of 4. The advantage of using the DNPH test stems from its ability to detect urinary branched-chain ketoacids in an outpatient setting. Standard urine test strips enable the detection of ketonuria. It offers an alternative source of testing in settings of restricted access to other diagnostic tests. Ketonuria is termed as a surrogate marker of underlying metabolic instability.[3] BCKAD enzyme activity can be measured in cells such as lymphoblasts, skin fibroblasts, and liver biopsy. In vivo measurements of BCKAD activity are not clinically useful because the activity levels do not correlate with leucine tolerance and oxidation.[3]
Molecular testing:
Molecular testing is available for three biallelic pathogenic gene variants. These include:
- BCKDHA gene: Encodes the E1-alpha subunit of the BCKAD enzyme complex (MSUD Type 1A).[3]
- BCKDHB gene: Encodes the E1-beta subunit of the BCKAD enzyme complex (MSUD Type 1B).[3]
- DBT gene: Encodes the E2 subunit of the BCKAD enzyme complex (MSUD Type 2).[3]
Genetic testing allows for a better understanding of the prognosis and genetic counseling of the family. Furthermore, it allows for accurate assessment of the deficient BCKAD subunit. This helps determine the individualized therapies. There are over 190 pathogenic variations in BCKAD enzyme subunits. All detected variants are homozygous or compound heterozygous
Diagnostic Strategies
Adult with symptoms of MSUD:
- DNPH testing can be used to alpha-ketoacids in the urine.[3]
- The most informative test is the identification of allo-isoleucine using plasma amino acid analysis.
- Branched-chain ketoacids and other organic acids can be detected using gas chromatography-mass spectrometry.
Newborn with signs and symptoms and a positive screening test for leucine, isoleucine, and valine or unexplained ketonuria:
- If infants are older than 48 to 72 hours, screening tests such as DNPH and urine ketone test strips can be used.
- Plasma amino acid analysis to detect elevated BCAA and allo-isoleucine.
- Gas chromatography-mass spectrometry is used to analyze the urine for ketoacids.
- Newborns and infants should not be challenged with higher than normal protein intake.[3]
Newborn with an affected sibling:
- If familial pathogenic variants are known, isolated blood from the umbilical cord can be used for a pathogenic variant detection by polymerase chain reaction (PCR), advanced sequencing, and melting analysis.
- If pathogenic variants are unknown, obtain blood from the umbilical cord to allow for pathogenic variant detection. The first diagnostic sign is the presence of maple syrup odor in the cerumen of a newborn. Therefore, inquire for a maple syrup smell 12 to 24 hours after birth. Allow protein intake to measure serum amino acid levels between 18 to 24 hours of life. If the results are vague, repeat the test between 24 to 36 hours of life. Amino acid profile indicative of MSUD should initiate dietary therapy. Furthermore, molecular testing and urine organic acid analysis should follow. It is important to remember that DNPH can't be used as a screening test until the age of 48 to 72 hours.
Treatment / Management
Effective treatment of maple syrup urine disease requires addressing the nutritional needs and optimally managing acute metabolic decompensations.[9] A pediatric nutritionist and metabolic disease specialist should be involved in the management of MSUD patients.
Medical Nutritional Therapy
The initiation of nutritional therapy requires clinical confirmation and a positive newborn screening result. The mainstay of treatment remains the dietary restriction of branched-chain amino acids. These dietary modifications need to be maintained throughout life. The goals of nutritional therapy are as follows [10]:
- Promote anabolism
- Prevent catabolism
- Promote normal growth and weight gain
- Preserve intellectual function
- Enable restriction of branched-chain amino acid in the diet, which helps reduce toxic metabolites
- Maintain plasma BCAA levels within the required treatment ranges
- Evaluate thiamine responsiveness
The allowed amounts of dietary BCAA are titrated into the diet using biochemical lab values and growth measurements during respective life periods. Long-term treatment warrants accurate assessment of caloric needs, BCAA restriction, BCAA-free amino acid supplementation, and valine and isoleucine supplementation.[2] BCAA-free medical foods can provide up to 80%-90% of protein needs. Valine and isoleucine help promote anabolism. They are supplemented because they are low in content in medical nutrition. Leucine supplementation is usually not required because they are found in ample amounts in breast milk or formula. Favorable intellectual outcomes can be achieved if leucine concentrations are maintained between 75 to 300 micromol/L.[11] It is also recommended that plasma valine and isoleucine levels are maintained between 200 to 400 micromol/L.[10] Thiamine should be supplemented at a dose of 50 to 200 mg/day in all patients with MSUD for four weeks. In thiamine responsive patients, thiamine supplementation should be continued. Thiamine should not be supplemented in patients diagnosed with the homozygous 1312T>A mutation or with mutations leading to less than 3% of BCKAD activity. Administration of sodium chloride may help maintain serum sodium concentrations and plasma osmolarity. It can help prevent the development of cerebral edema or fatal herniation.
Treatment of Acute Metabolic Decompensation
Metabolic decompensations (plasma leucine >380 micromol/L) usually occur due to dietary non-compliance and infections. Dietary non-compliance raises the BCAA levels and rarely progresses to decompensation and encephalopathy. However, trauma and infections can trigger large protein catabolism leading to a metabolic crisis. Decompensations arise more commonly in the first year of life and after the age of 15.[12] Strauss et al. indicated that vomiting and viral gastroenteritis are the most common cause of hospitalization during acute decompensation.[13] Other common causes of hospitalization include viral bronchiolitis, sinusitis, neonatal encephalopathy, and urinary tract infections.[13] Residual BCKAD activity and liberation of leucine from catabolism determine the risk of metabolic crisis.[3] Patients with a higher residual BCKAD activity have a better leucine tolerance when well. Furthermore, during an illness, these patients face less severe elevations in leucine. The main aim of therapy is to suppress protein catabolism and promote protein synthesis.[2] Management strategies in more severe cases include:
- Effectively treating the underlying stressor causing the metabolic crisis
- Restrict protein intake for 24 to 72 hours
- Provide ample caloric support
- Provide adequate hydration to maintain metabolic homeostasis
- Provide supplementation with cofactors
- Eliminate toxic metabolites
- Treat associated clinical sequelae
- Correct metabolic abnormalities
Home Therapy
Providers can be instructed to use a dinitrophenylhydrazine reagent to detect high urine branched-chain ketoacids. This allows for the timely detection and home management of mild to moderate cases of acute metabolic decompensation. In such cases, experienced providers can help manage with restriction of dietary leucine, sick day formulas, and outpatient monitoring.[3] More specifically, sick day protocols require a 120% increase in the intake of BCAA-free amino acid formula, fluid administration of 150 mL/kg, 50% to 100% decrease in leucine intake, and frequent small feedings.[2] It is important to ensure that nutritional treatment is aggressive and provides sufficient energy.
In-Hospital Therapy[3]
Goals and treatment strategies of in-hospital therapy include:
- Effectively treating the underlying stressor (e.g., fever, dehydration, infection, and inflammation).
- To control nausea and vomiting, antiemetics such as ondansetron should be administered.
- Reduction in leucine concentration at 750 micromol/L or more per 24 hrs. This reduction in leucine can be achieved via insulin and glucose infusions. Ideally, leucine levels should be maintained from 200 to 300 micromol/L. Upon clinical improvement, total parenteral nutrition can be used to reintroduce protein back into the diet (25%-50% of normal intake). This intake can be increased depending on the clinical situation over the next few days.
- EER must be provided at least 1.25 times the weight or surface area. Lipid should constitute 40% to 50% of total calories. Nutritional goals can be achieved by combined parenteral and enteral feeding.
- Isoleucine and valine should be supplemented at 20-120 mg/kg/day each. The intake of supplements is adjusted to maintain a steady plasma concentration of 400-600 micromol/L.
- Enteral supplementation of tyrosine (100-400 mg/kg/day) to treat focal or generalized dystonias.
- Glutamine and alanine supplementation at 150-400 mg/kg/day each.
- Supplement BCAA-free amino acids.
- Maintain sodium levels within the physiological range.
- Correct underlying acid-base disturbances.
- Avoid osmolarity fluctuations > 5 mosm/L per day and maintain urine output.
- Prevent and treat hypokalemia and hypophosphatemia associated with IV glucose and insulin therapy.
Role of Sodium Phenylbutyrate (NaPBA)
Sodium phenylbutyrate is a nitrogen scavenger. It is primarily used in the treatment of urea cycle disorders. NaPBA can also reduce branched-chain amino acid levels. It can be used in patients with intermediate MSUD.[14] Hemodialysis and peritoneal dialysis can also be used to rapidly correct BCAA and ketoacids during an acute decompensation.
Orthotopic Liver Transplantation
Indications:
- Psychomotor disabilities
- Poor metabolic control
- Frequent metabolic decompensations
The liver is responsible for expressing 10% of BCKAD activity. Therefore, liver transplantation is recommended for classic MSUD patients who are difficult to manage. More commonly, the liver from an undeceased and unrelated individual is used.[2] Post-transplantation, the residual activity of BCKAD, can rise to that of mild MSUD levels. Therefore, transplantation tends to reduce the requirement of dietary restrictions and episodes of metabolic decompensation. It also does not reverse previously inflicted brain damage, cognitive dysfunction, and psychiatric illnesses.[15] However, transplantation doest restrict further progression of neurological impairment and prevent the development of cerebral edema.[16][17]
Management in Pregnancy
It is indeed possible for women with MSUD to deliver a healthy child. The mother must be educated about the potential teratogenic risks of elevated maternal leucine concentration. In such patients, tight metabolic control before and throughout gestation is critical. As the placenta and fetus develop, the maternal need for protein and BCAA exponentially increases. Therefore, measurements of plasma amino acid concentrations and fetal growth are imperative to avoid possible nutritional deficiencies. Maintaining BCAA levels between 100 and 300 micromol/L is said to be compatible with normal infant delivery. At the time of delivery, referral to a metabolic center must be made. This is because the post-partum period is deemed dangerous for the mother. Events such as labor stress, internal blood sequestration, and uterine involution can act as a source of metabolic decompensation. Therefore, extra measures must be taken to counteract catabolism during the post-partum period.
Management of other MSUD Complications
Cerebral Edema
- Administer furosemide (0.5 to 1.0 mg/kg)
- Infusion of mannitol (0.5 to 1.0 g/kg) followed by hypertonic saline (3%-5%)
The risk of developing cerebral edema can be reduced by elevating the head of the bed.[3] Furthermore, it is recommended to use a PICC line in encephalopathic patients. The use of a PICC line allows the delivery of nutrition without the need for excess fluid volume.[3]
Cerebral Herniation
- Elevate the head
- Induce hyperventilation with the help of a face mask or endotracheal tube.
- Infuse mannitol and hypertonic saline.
- Transfer the patient immediately to a pediatric/neonatal intensive care unit.
Infection
Patients with MSUD are predisposed to candida infections and catheter-based bacterial or fungal infections. It is vital to monitor patients for hospital-acquired infections continuously. Failure to treat them can result in an episode of acute metabolic decompensation.
Acute Pancreatitis
During an acute metabolic decompensation episode, a patient may develop symptoms such as epigastric pain, mid-back pain, anorexia, and vomiting. This should raise suspicion of acute pancreatitis and immediately order serum levels for lipase and amylase. Also, the physician must suspend enteral feeding and keep the patient NPO. Treatment is usually supportive, and the patient's nutritional needs can be managed with the use of special parenteral solutions.
Neuropsychiatric Illnesses
Adult and adolescent patients are at an increased risk of developing anxiety, depression, and ADHD. These can be treated by prescribing standard anti-depressants and psychostimulant drugs.
Secondary Complications
Surgical procedures and trauma care should be planned in coordination with a metabolic specialist.
General Prevention
Patients must be advised never to exceed their daily allowed dose of branched-chain amino acids.
Differential Diagnosis
It is essential to exclude other clinically distinct entities that can also manifest as neonatal encephalopathy. These include:
- Hypoglycemia
- Status epilepticus
- Meningitis
- Encephalitis
- Kernicterus
- Birth asphyxia
- Urea cycle defects[3]
- Organic acidopathies such as propionic acidemia and methylmalonic acidemia[3]
- HMG-CoA lyase deficiency
- Beta-ketothiolase deficiency[3]
- Non-ketotic hyperglycinemia[3]
Sotolone, which is found in fenugreek and lovage, is responsible for the characteristic maple syrup odor in cerumen and bodily secretions.[3] Excess ingestion of fenugreek during pregnancy can result in a false diagnosis of MSUD.[3] In the setting of NICUs, the use of topical benzoin can also give off a sweet odor.[3]
Prognosis
Good prognosis can be expected in patients who begin therapy before or immediately after developing symptoms. Plasma leucine concentrations are known to affect neurocognitive outcomes. Classic MSUD in school-aged patients has shown high performance than IQ.[18] 61% of adult MSUD patients live independently and integrate well into society.[12] However, 56% of patients still require psychological and psychiatric care.[12] Patients with late-onset MSUD can suffer from slight developmental delays depending on the residual BCKAD activity.[3] A delayed diagnosis of more than 7 to 14 days of classic MSUD can result in irreversible learning disability and cerebral palsy.[3]
Complications
Failure to diagnose and treat MSUD in a time-sensitive manner can result in serious consequences. Patients undergoing a treatment plan can develop an acute illness resulting in a sudden increase in levels of branched-chain amino acids. This metabolic crisis is usually indicated by the development of clinical symptoms such as extreme fatigue, irritability, vomiting, and loss of alertness. If the patient remains undiagnosed or untreated, the following complications can arise:
- Seizures
- Metabolic acidosis
- Cerebral edema
- Cerebrovascular ischemia
- Intellectual disabilities
- Blindness
- Muscle spasticity
- Irreversible neurological damage
- Osteoporosis
- Acute pancreatitis
- Recurrent esophageal candidiasis due to T-cell suppression from elevated plasma leucine.
- Essential amino acid deficiency presenting as anemia, hair loss, growth failure, and acrodermatitis
Deterrence and Patient Education
A thorough understanding of the patient and the public's clinical condition will ensure adherence to optimal patient care strategies. Furthermore, it allows physicians and other healthcare team professionals to provide the best patient outcomes based on evidence-based medicine. Educational resources should include:
- Educational handouts obtained online from the New England Consortium of metabolic programs
- The American College of Pediatrics website
- MSUD family support group
- National Library of medicine genetics home reference
- NCBI genes and diseases
- Metabolic Support UK
- European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD)
- Organic Acidemia Association
Enhancing Healthcare Team Outcomes
Optimal management of maple syrup urine disease requires an interprofessional team consisting of an internist, pediatrician, geneticist, metabolic specialist, pediatric nutritionist, social worker, and pediatric nurse. The mainstay of therapy is medical, nutritional therapy. The healthcare team must collaborate to assess caloric needs and execute dietary treatment strategies such as BCAA restriction, BCAA-free amino acid supplementation, and valine and isoleucine supplementation. The pediatric nutritionist must educate the patient never to exceed the daily intake of amino acids. They should also educate the patient to adhere to a lifelong restricted diet. The geneticist must counsel the patient or their parents about their inherited disorder's implications to help them make informed medical and personal decisions. Outcomes of the patient depend upon biochemical findings, clinical severity, and molecular testing. A combination of early diagnosis, aggressive treatment, and controlling plasma leucine levels help improve neurological outcomes.[19]
(Click Image to Enlarge)
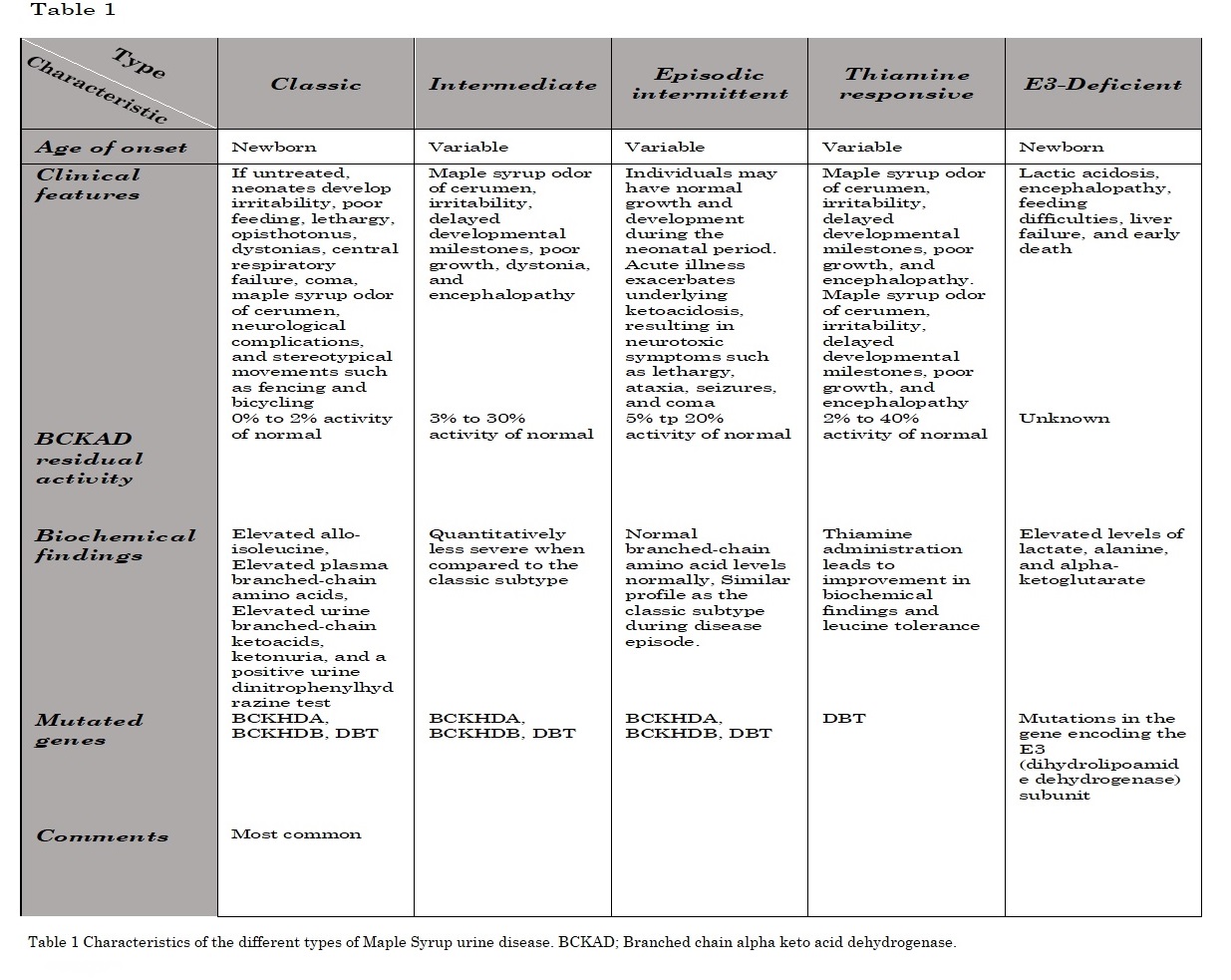
Table 1 Characteristics of the different types of Maple Syrup urine disease
Contributed by Mahdi Alsaleem, MD